ACESSO REMOTO A SERVIDORES E INTRODUÇÃO AO LINUX
Sistema
operacional
Em
Bioinformática é comum o uso de computadores que utilizam sistemas operacionais
baseados em Linux [1]. Os sistemas operacionais relacionados abaixo são todos
baseados nessa plataforma e possuem acesso gratuito:
· CentOS (distribuição gratuita do RedHat Enterprise) [2]
· Fedora [3]
· Ubuntu [4]
A distribuição CentOS é indicada para servidores.
Como se
conectar a partir de Windows
É possível acessar servidores através de uma máquina que possui como sistema operacional o Windows.
· Para troca de arquivos: Secure Shell [5]
· Conexão direta SSH: Putty [6].
ssh usuario@maquina.icb.ufmg.br
Para
trabalhar com Linux, alguns comandos de uso rotineiro precisam ser aprendidos.
A seguir, uma lista com alguns comandos básicos e uma breve descrição:
·
cp teste.seq manuel
Copia o
arquivo teste.seq, que se encontra dentro do
diretório corrente, para o diretório Manuel, também dentro do diretório
corrente.
·
cp teste.seq ../manuel
Nesse
caso, o diretório manuel está em um nível acima do
diretório corrente.
·
cp /home/user/readme.txt . (note o “ponto”
separado por espaço)
Copia o arquivo que está no caminho especificado para o diretório
corrente. Note que o ponto final ao término do comando é utilizado para indicar
diretório corrente.
·
mv teste.seq manuel/seqs
Move o
arquivo teste.seq para o diretório seqs, o qual está dentro de manuel
·
mv teste.seq teste.fasta
Como o
diretório de destino para o arquivo teste.seq é o
mesmo de origem, o arquivo é renomeado e seu conteúdo mantido inalterado.
Usa-se para trocar o nome de um arquivo, na verdade.
·
mkdir seqs
Cria o
diretório denominado seqs.
·
rmdir seqs
Remove o
diretório seqs quando ele se encontra vazio.
·
rm teste.seq
Remove o
arquivo teste.seq.
·
cd ..
Muda-se
do diretório corrente para o diretório acima.
·
cd manuel
Entra no
diretório denominado manuel.
·
ls
Lista
conteúdo do diretório corrente.
·
ls -l
Lista o
diretório corrente com mais informações.
·
man ls
Mostra o
manual para o comando ls.
·
q
Interrompe
a listagem do output do manual e do comando
<more> do Linux.
·
more readme.txt
No Linux
imprime na tela o conteúdo do arquivo, nesse caso, readme.txt
·
head readme.txt
Imprime
as primeiras linhas do arquivo.
·
tail readme.txt
Imprime
as últimas linhas do arquivo.
Tabulador:
são usados para nomes compridos. O tabulador completa o nome que esta sendo
digitado desde que o mesmo exista no diretório corrente. Recomenda-se
fortemente usar o tabulador ao digitar caminhos compridos como
·
more /home/manuel/seqs/readme.txt
pois se
a máquina completa o nome, é porque o caminho está sendo digitado corretamente.
Use sempre!
Asterisco:
utilizado para representar qualquer caractere, por exemplo:
·
more *txt
Imprime
na tela o conteúdo de readme.txt, assim como
·
more *dme*.
Acessando uma conta em um servidor
Abra o programa Putty.
Complete o campo Host Name com o IP do servidor e mude Connection Type ou Protocol para a opção SSH.
Complete o campo Saved Sessions com o nome desejado.
Clique o botão Save.
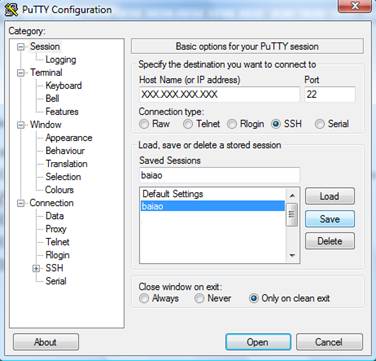
Figura 1: Tela inicial do programa Putty.
De agora em diante com um duplo clique no nome escolhido o terminal abrirá automaticamente.
Acesse o conteúdo
disponível em: http://biodados.icb.ufmg.br/tutorial2011/Linux/,
ou utilize arquivos próprios para executar os comandos a seguir em um terminal
Linux.
· mkdir treinamento
· cd treinamento [copie todo o conteúdo do link acima para esta pasta]
· ls
Veja o conteúdo do arquivo
· more aldolase.cds
Para editar um arquivo utilize o editor de texto denominado vi
· vi aldolase.cds
Para entrar no modo de inserção digite a letra “i”. Observe que no canto inferior esquerdo da tela aparece a palavra INSERT. Nesse momento você é capaz de digitar livremente. Movimente o cursor com as setas do teclado.
Troque o nome e descrição da seqüência FASTA para apenas >teste
Pra salvar a modificação tecle ESC, em seguida dois pontos (:) e por fim x! seguido de ENTER.
Veja como ficou o arquivo após a edição
· more aldolase.seq
Pratique com outras seqüências presentes no mais conhecido banco de dados biológico do mundo, o National Center for Biotechnology Information, ou simplesmente NCBI [7].
Por exemplo, procure por myoglobin no NCBI, utilizando o banco de dados protéico, denominado Protein, como mostrado na Figura 2 abaixo:
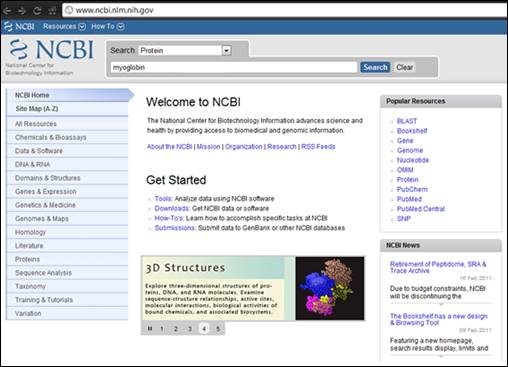
Figura 2: Website do
Escolha uma seqüência proveniente do resultado da busca e então selecione
a opção Send to File e abra o arquivo.
Digite em seu terminal:
· vi mioglobina.pep
Selecione com o mouse o conteúdo da seqüência e copie.
Clique no terminal com o botão direito do mouse e a seqüência será colada.
Salve o novo arquivo gerado como antes (ESC : x!)
OBS: Consulte o manual para mais informações sobre seus comandos (man vi).
Consultando
o NCBI
Uma consulta simples como a exemplificada na Figura 2 geralmente retorna uma quantidade numerosa de dados o que pode ser um complicador para obtenção dos dados desejados.
Para filtrar uma consulta, existem diferentes recursos que podem ser adicionados que ajudam a limitar e direcionar o resultado. Por exemplo, as consultas abaixo combinam palavras chaves contidas entre colchetes e operadores lógicos (AND, OR e NOT) para direcionar a busca.
·
Einstein[Author] AND relativity
theory[Title]
·
PGM1[Gene Name] AND Homo
sapiens[Organism] OR Mus musculus
NOT fragment
Aprendendo
um novo comando, grep
Observe o arquivo db.fasta
· more db.fasta (aperte “q” para sair)
Para descobrir quantas seqüências protéicas
existem nesse arquivo poderíamos simplesmente contar, o que seria uma tarefa
fácil em arquivo pequeno como esse. Após a contagem descobrimos que existem 16
seqüências. Mas se pensarmos em um arquivo contendo todas as proteínas humanas,
essa tarefa já seria inviável. Para simplificar, podemos utilizar o comando grep, que
identifica rapidamente linhas que possuem os caracteres especificados dentro de
um arquivo. Como cada seqüência protéica apresentada
· grep “>” db.fasta –c
A opção –c é utilizada para especificar que desejamos apenas a contagem final. Caso essa opção fosse retirada, o usuário observaria uma listagem das linhas que contem o conteúdo especificado dentro das aspas. Teste sem o –c.
Pronto agora você já sabe contar rapidamente seqüências em um arquivo.
Banco de dados de seqüências e busca de similaridade BLAST no NCBI
Retorne à página do NCBI e procure pela ferramenta BLAST [7, 8 e 9] (Basic Local Alignment Search Tool). Essa ferramenta é muito utilizada em Bioinformática para pesquisa de similaridade entre seqüências biológicas, sejam elas de nucleotídeos ou de aminoácidos.
Clique
Altere a Database
para Others (nucleotide collection nr/nt)
Altere o algoritmo
O MEGABLAST é uma versão que deve ser usada para, por exemplo, identificar uma seqüência humana no próprio genoma humano e é muito mais rápido que o BLASTn tradicional. Mas para iniciar a busca ele requer encontrar uma sequencia idêntica maior que o BLASTn exige.
A versão “descontínua” do MEGABLAST (discontiguos MEGABLAST) utiliza uma janela de início de alinhamento degenerada na terceira posição para favorecer o início do alinhamento quando códons variando na terceira base ocorrem (alinhamentos de sequencias de organismos muito distantes).
Digite cinco (5) linhas de bases nucleotídicas (ACTG) aleatórias no formato fasta como exemplificado abaixo.
>seunome
acgatcgatcgatcgatcgatcgtagctacgtacg...
Logo após clique
Funcionamento
do BLAST e valor de E-value
O algoritmo do BLAST fragmenta sua seqüência (denominada query) em pedaços (ou seed) de tamanho W. Encontra na Database os pareamentos PERFEITOS da seed. A seed não pode ser muito pequena para não iniciar pesquisas demais e de pouca importância.
O algoritmo “caminha” para as extremidades tentando estender o alinhamento o máximo possível; nesse processo podem ser inseridos gaps, que são pequenas falhas nos alinhamentos.
A pontuação para cada passo do alinhamento é dada segundo uma matriz de pontuação. Bases idênticas resultam em pontuação positiva enquanto abrir gaps, estender gaps e bases diferentes (mismatches) são penalizadas com pontuação negativa.
O algoritmo termina quando estender mais “não compensar”, logo, determina a máxima subseqüência alvo (subject) que retorna o maior escore dentro da Database.
Assim, o programa visa detectar a seqüência mais similar baseado em subseqüência máxima (alinhamento local), o que é um grande facilitador para bioinformatas por se tratar de um processo computacionalmente mais rápido.
A questão da homologia relaciona-se com o cálculo de E-value: quantos alinhamentos iguais ou melhores que este que você observa poderiam ser obtidos sem nenhuma relação de homologia entre as seqüências ai alinhadas? Se for um número improvável, você pode resolver desconsiderar a hipótese nula (não homologia) não pode? Portanto, quanto MENOR, mais próximo de zero for o valor de E-value obtido no experimento com bases aleatórias acima, melhor. Diferentes experimentos consideram diferentes valores, variando geralmente entre 1 x 10-5 e 1 x 10-15.
Formatando a Database para uso do BLAST em sua máquina
O BLAST necessita de databases formatadas. Para isso existe o comando formatdb.
formatdb –i db.fasta –o T –p F
i: input
o: Indexa os dados que aparecem no FASTA.
p: Responde à seguinte pergunta: É proteína? Caso não: Falso, caso sim True.
O significado de alguns parâmetros BLAST
p: Programa: tblastn, etc.
i: Entrada: query.
d: Database: tem que estar formatada.
e: E-value: o número de alinhamentos iguais ou melhores que podem ser obtido sem homologia.
F: Liga ou desliga o filtro de baixa complexidade.
b: O número de alinhamentos que serão reportados para cada query.
a: Número de processadores que serão utilizados.
m: É o formato do arquivo de saída.
o: É o nome que você quer para o arquivo de saída.
Referências
[1] Linux: http://br-linux.org/
[2] CentOS: http://www.centos.org/
[3] Fedora: http://fedoraproject.org/pt/
[4] Ubuntu: http://www.ubuntu-br.org/
[5] SSH Secure Shell: http://cromatina.icb.ufmg.br/ssh/
[6] Putty: http://www.putty.org/
[7] NCBI: http://www.ncbi.nlm.nih.gov/
[8] Tutorial em inglês para uso da ferramenta BLAST: http://www.ncbi.nlm.nih.gov/books/NBK21097/
[9] Stephen F. Altschul, Warren Gish, Webb Miller, Eugene W. Myers and
David J. Lipmanl: Basic Local Alignment Search Tool. J. Mol. Bio (1990) 215, 403-410.