Análise de transcriptoma
usando a base de dados Kegg Orthology
$megablast -i h.sapiens.nuc –d
tumor.seq –D 3 –F F –a 4 –p 96 –s 100 –o megakegg
-i = input
-d = database
-D 3 = saida
tabulada
-F F = filtro de baixa complexidade
desligado (F = False)
-a 4 = use 4 processadores
-p 96
= mínima identidade 96%
(seqüenciamento pode ter até 4% de erro)
-s
100 = mínimo escore 100
-o = nome do arquivo de saída
$cat saída | awk ‘{print $1}’ | sort | uniq –c | sort –k 1,1 –n –r >
resultado
awk =
nenhuma condição (aspas) e ação de imprimir coluna 1 (chaves)
sort =
ordena as queries (já estão ordenadas, só pra garantir)
uniq =
mostra cada query uma única vez
uniq –c =
idem, mas mostra quantas de cada haviam
sort –k 1,1 =
ordena pela coluna 1, que é o número de ocorrências das queries
sort –n =
entende valores como números
sort –r = reverso, ordena do maior para o
menor
Selecione
algum hit e procure a identificação no arquivo h.sapiens.nuc
$ cat h.sapiens,nuc | grep “hsa:1234”
Há como fazer de forma
global usando banco de dados (MySQL)
Para isso é necessário
preparar os arquivos e tabelas a serem utilizadas. Bora
aprender um pouco de MySQL?
1) ls /home/bacharelado/mysql_aula
2) No seu /home/SEU_NOME/ crie um novo diretorio:
mkdir mysql_aula
3) cd mysql_aula
4) cp /home/bacharelado/mysql_aula/*
. {opag}
5) Crie um arquivo tabulado com apenas algumas colunas do
blast:
cat megakegg | awk -v
OFS="\t" '($1 != "#") {print
$1,$2,$3,$11,$12}' > megakegg_tab
-v OFS à insere um tabulador entre as colunas selecionadas
6) Nessa mesma pasta, onde estão os arquivos, acesse o MySQL com seu username e senha (ambos iguais ao nome do diretorio do usuario).
mysql -u
<username> -p <ENTER>
7) Informe a senha
8) Listar bancos de dados disponíveis:
show databases;
9) Informe ao mysql qual banco de
dados vai usar (banco de dados tem o mesmo nome do usuario)
use <username>;
10) Verifique que o banco ainda está vazio (sem tabelas):
show tables;
VEJA O PROCEDIMENTO
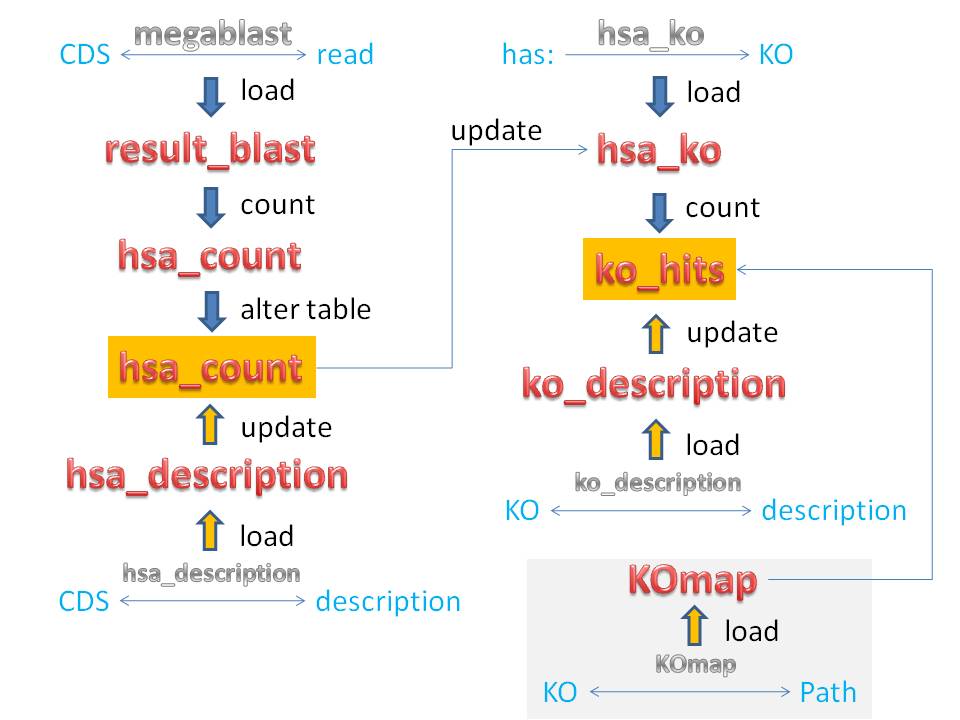
11) Crie uma tabela para armazenar os dados selecionados
do BLAST:
create table result_blast (cds varchar(15), subject varchar(50), identity
double(5,2), evalue varchar(10),
score int, index cds_idx (cds));
12)
Verifique a estrutura da tabela criada:
desc result_blast;
13) Carregue o resultado do blast
para a tabela:
load data local infile 'megakegg_tab' into table result_blast;
14) Verifique a tabela criada:
select * from result_blast limit
10;
Veja que o mesmo arquivo agora se encontra em colunas no
banco de dados
15) a consulta anterior utilizando awk
|sort|uniq já no informou quais hsas
deram mais hits e portanto estão mais presentes nessa amostra de tumor. Que tal
utilizar o banco pra isso?:
select *, count(*) from result_blast group
by cds limit 10;
- mais simples né?
16) Crie uma tabela dessa contagem de hsas:
create table hsa_count select cds, count(*) as hits from result_blast group
by cds;
17) Verifique a tabela criada:
select * from hsa_count limit
10;
18) Cansado de ver apenas símbolos, sem saber o que eles
são? Crie uma tabela contendo descrições dos genes:
create table hsa_description (cds varchar(15),
description varchar(150), index cds_idx (cds));
19) Verifique a tabela criada, sempre bom:
desc hsa_description;
20) Carregue as descrições na tabela recém criada:
load data local infile 'hsa_description' into table hsa_description;
21) Verifique os dados carregados, sempre bom também, vai
que ta errado:
select * from hsa_description limit
10;
22) Agora altere a primeira tabela hsa_count
para conter tambem a coluna de descricao
do gene:
alter table hsa_count add
column description varchar(150);
23) Visualize nova estrutura da tabela, ta bom.. so se quiser:
desc hsa_count;
24) Atualize a tabela hsa_count
com dados de descricao de genes:
update hsa_count, hsa_description set hsa_count.description = hsa_description.description where hsa_count.cds = hsa_description.cds;
- tranqüilo?
25) Verifique a tabela agora com dados novos:
select * from hsa_count limit
10;
- ahhh, agora sim!
26) Qual o nome do carinha que da mais hit mesmo?
select * from hsa_count order
by hits desc limit 10;
OK, você é quase-biólogo e não sabe por que esta criando
tabelas a essa hora da manha? Então com base nesses dados, formule uma hipótese
de como obter a cura desse câncer.
27) Agora vamos relacionar nossos hsas
grupos KO? Miguel, o que é KO mesmo?
Crie uma tabela que relaciona KOs
e cds:
create table hsa_ko (cds varchar(15),
ko varchar(11), hits bigint default 0, index cds_idx (cds), index ko_idx(ko));
desc hsa_ko;
28) Perdido? Não sabe quantas tabelas já criou?
show tables;
29) Carregue os dados na tabela recém criada:
load data local infile 'hsa_ko.list' into table hsa_ko;
30) Inclua a contagem de CDS na tabela recém criada:
update hsa_ko, hsa_count set hsa_ko.hits = hsa_count.hits where hsa_ko.cds = hsa_count.cds;
31) Verifique o numero de pares hsa
x ko (inclusive os CDS que não tiveram hits):
select count(*) from hsa_ko;
32) Vamos excluir pares hsa x ko cujo CDS não obtiveram hits e não nos interessa:
delete from hsa_ko where
hits = 0;
33) Verifique o numero de pares hsa
x ko restantes:
select count(*) from hsa_ko;
Muito menos né, claro, nem
todos hsas estão presentes na amostra
34) Qual KO danadinho deu mais hit?
select * from hsa_ko order
by hits desc limit 10;
35) Agora crie mais uma tabela contendo o numero de cds e o total de hits para cada ko.
create table ko_hits select ko, count(distinct cds) as total_cds, sum(hits) as total_hits
from hsa_ko group by ko;
36) Verifica tabela criada:
select * from ko_hits limit 10;
37) Já sei, mais uma vez cansado de ver identificadores sem
saber o que eles são neh. Então crie maaaais uma tabela com descrições de KO:
create table ko_description (ko varchar(11)
primary key, description varchar(150));
38) Popule (em mysqelês)
essa tabela:
load data local infile 'ko_desc' into table ko_description;
39) Verifique os dados:
select * from ko_description limit 10;
40) Adicione coluna de descricao
do ko na tabela ko_hits, ta
lembrado como neh
alter table ko_hits add
column ko_desc varchar(150);
41) Atualize a tabela ko_hits
com as descricoes:
update ko_hits, ko_description set ko_hits.ko_desc = ko_description.description where ko_hits.ko = ko_description.ko;
42) Dá uma oiadinha:
select * from ko_hits limit 10;
43) qual KO tem mais hits?
select * from ko_hits order
by total_hits desc limit
10;
44) Pergunte ao Miguelito um KO legal. Efetue a busca
LIKE na tabela de KOs:
select * from ko_hits where ko_desc
like '%o que o miguelito disse%';
ou
select * from ko_hits where ko_desc like '%protease%';
45) Pra finalizar, crie uma tabela que relaciona KOs e vias metabólicas, por que não?:
create table KOmap (path varchar(25), ko varchar(25), path_desc varchar(150));
46) Popule a tabela:
load data
local infile 'KO2map'
into table KOmap;
47) Agora é quebradera, já
sabemos usar select,
create, show, load, alter, order by, update, limit, where, like, count ... Tudo que um biólogo precisa saber certo? Errado, falta o
JOIN!
Misture dados de tabelas
distintas para facilitar a observação dos resultados:
Aproveita, e mistura vários
comandos vai... select, join, order by
e limit
select ko_hits.*, KOmap.path, KOmap.path_desc from
ko_hits inner join
KOmap on ko_hits.ko = KOmap.ko order
by total_hits desc limit 10;
Observe a redundância, um KO
pertence a diferentes vias metabólicas e elas podem ser visualizadas nessa
consulta.
APRENDEU A “PERGUNTAR BEM”???
{kind=link}