
Science, Vol 300, Issue 5618,
489-492 , 18 April 2003
[DOI: 10.1126/science.1083558]
Induction of Tumors in Mice by Genomic Hypomethylation
François
Gaudet,1,2,3 J. Graeme
Hodgson,4 Amir Eden,1
Laurie Jackson-Grusby,1 Jessica
Dausman,1 Joe W. Gray,4
Heinrich Leonhardt,2,3
Rudolf Jaenisch1*
Genome-wide DNA hypomethylation occurs in many human
cancers, but whether this epigenetic change is a cause or
consequence of tumorigenesis has been unclear. To explore
this phenomenon, we generated mice carrying a hypomorphic
DNA methyltransferase 1 (Dnmt1) allele, which
reduces Dnmt1 expression to 10% of wild-type levels and
results in substantial genome-wide hypomethylation in all
tissues. The mutant mice were runted at birth, and at 4
to 8 months of age they developed aggressive T cell lymphomas
that displayed a high frequency of chromosome 15 trisomy.
These results indicate that DNA hypomethylation plays a
causal role in tumor formation, possibly by promoting
chromosomal instability.
1 Whitehead Institute for Biomedical Research and
Department of Biology, Massachusetts Institute of Technology,
Cambridge, MA 02142, USA.
2 Ludwig Maximilians
University, Department of Biology II, 80336 Munich,
Germany.
3 Max Delbrück Center for Molecular Medicine,
13125 Berlin, Germany.
4 Department of Laboratory
Medicine and UCSF Comprehensive Cancer Center, University of
California, 2340 Sutter Street, San Francisco, CA 94143, USA.
* To whom correspondence
should be addressed. E-mail: jaenisch@wi.mit.edu
Human cancer cells often display abnormal patterns of DNA
methylation. The role of aberrant hypermethylation in the
silencing of tumor suppressor genes is now well
documented (1).
In contrast, the role of aberrant hypomethylation—which
is observed in a wide variety of tumors (2–5),
often together with regional hypermethylation—has
remained unclear.
To investigate whether DNA hypomethylation has a causal role
in tumor formation, we generated mice with highly reduced
levels of Dnmt1, the enzyme that maintains DNA
methylation patterns in somatic cells (6).
Because mice homozygous for a Dnmt1 null allele
(Dnmt1c/c) die during gestation (7,
8),
we combined a hypomorphic allele
[Dnmt1chip (9)]
with a null allele to generate
Dnmt1chip/c (referred to here as
Dnmt1chip/–) compound heterozygotes
with a substantially reduced level of genome-wide DNA
methylation. Dnmt1chip/– embryonic stem (ES)
cells expressed 10% of wild-type levels (Fig.
1A). To test whether the reduced Dnmt1 expression
affected DNA methylation in vivo, we generated mice
carrying the different Dnmt1 alleles and determined
their global methylation levels with the use of a probe
for endogenous retroviral A type particles (IAPs) (Fig.
1, B and D) (10)
and centromeric repeats (Fig.
1C). Southern blot analysis of embryonic fibroblasts
and adult tissues showed that the DNA from compound
heterozygotes was hypomethylated relative to the DNA from
Dnmt1chip/chip or Dnmt1+/+ mice,
although substantially less so than the DNA from
Dnmt1–/– null ES cells. Mice carrying
the different Dnmt1 alleles were obtained at the
expected Mendelian ratios, indicating that reduction of
Dnmt1 expression to 10% was compatible with viability.
However, compound heterozygotes
(Dnmt1chip/–) were runted and their weight
at birth was only 70% that of Dnmt1+/– mice,
in contrast to mice homozygous for the hypomorphic allele
(Dnmt1chip/chip), which were normal in
size (Fig.
2A). Dnmt1chip/– mice, although
remaining substantially underweight, were fertile and
generated litters of nonrunted pups when bred with
wild-type mice.
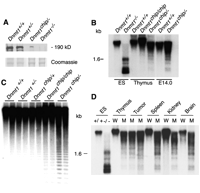 Fig. 1. Genomic
hypomethylation in Dnmt1 hypomorphic mice. (A)
Immunoblot analysis of protein extracts from ES cells, using a
C-terminal Dnmt1 chicken antibody (34).
The level of Dnmt1 in Dnmt1chip/– ES cells
was markedly reduced, demonstrating the hypomorphic nature of
this allele. The Dnmt1–/– negative control
extracts did not show any bands, as expected [c allele (7)].
A Promega antibody to chicken immunoglobulin Y (IgY), with
horseradish peroxidase as secondary antibody, was used;
detection was performed with the Amersham Pharmacia Biotech
ECL kit. The Coomassie gel shows total protein levels in each
sample. (B) Southern blot of methylation status of IAPs
in Dnmt1chip/chip and
Dnmt1chip/– mice. Genomic DNA was digested
with the methylation-sensitive restriction enzyme Hpa II and
probed with an IAP cDNA probe (10).
Hypomethylation was detected in thymus from 6-week-old
Dnmt1chip/– mice and in
Dnmt1chip/– embryonic day (E) 14.0
fibroblasts but not in Dnmt1chip/chip
homozygotes or wild-type controls, as evidenced by the
presence of lower molecular weight DNA fragments. (C)
Southern blot analysis of centromeric repeat methylation of
E14.0 fibroblasts from mice containing various Dnmt1
alleles. Hypomethylation is evident in the
Dnmt1chip/– lanes. (D) Southern blot
analysis of IAP methylation status in tissues from a
6-week-old Dnmt1chip/– mouse and in
lymphomas. Significant hypomethylation is evident in all
Dnmt1chip/– tissues. Controls for
methylation
(Dnmt1+/+ ES
cells) and hypomethylation (Dnmt1–/– ES
cells) are also shown (ES, ES cells; W,
Dnmt1+/+;M,
Dnmt1chip/–). [View
Larger Version of this Image (81K GIF file)] Fig. 1. Genomic
hypomethylation in Dnmt1 hypomorphic mice. (A)
Immunoblot analysis of protein extracts from ES cells, using a
C-terminal Dnmt1 chicken antibody (34).
The level of Dnmt1 in Dnmt1chip/– ES cells
was markedly reduced, demonstrating the hypomorphic nature of
this allele. The Dnmt1–/– negative control
extracts did not show any bands, as expected [c allele (7)].
A Promega antibody to chicken immunoglobulin Y (IgY), with
horseradish peroxidase as secondary antibody, was used;
detection was performed with the Amersham Pharmacia Biotech
ECL kit. The Coomassie gel shows total protein levels in each
sample. (B) Southern blot of methylation status of IAPs
in Dnmt1chip/chip and
Dnmt1chip/– mice. Genomic DNA was digested
with the methylation-sensitive restriction enzyme Hpa II and
probed with an IAP cDNA probe (10).
Hypomethylation was detected in thymus from 6-week-old
Dnmt1chip/– mice and in
Dnmt1chip/– embryonic day (E) 14.0
fibroblasts but not in Dnmt1chip/chip
homozygotes or wild-type controls, as evidenced by the
presence of lower molecular weight DNA fragments. (C)
Southern blot analysis of centromeric repeat methylation of
E14.0 fibroblasts from mice containing various Dnmt1
alleles. Hypomethylation is evident in the
Dnmt1chip/– lanes. (D) Southern blot
analysis of IAP methylation status in tissues from a
6-week-old Dnmt1chip/– mouse and in
lymphomas. Significant hypomethylation is evident in all
Dnmt1chip/– tissues. Controls for
methylation
(Dnmt1+/+ ES
cells) and hypomethylation (Dnmt1–/– ES
cells) are also shown (ES, ES cells; W,
Dnmt1+/+;M,
Dnmt1chip/–). [View
Larger Version of this Image (81K GIF file)]
|
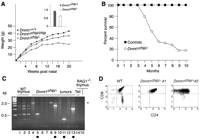 Fig. 2. Dnmt1
hypomorphs are runted and develop T cell lymphomas. (A)
Average weight of Dnmt1chip/+
(n = 20) and Dnmt1chip/– (n =
20) male littermates at birth (inset).
Dnmt1chip/– mice were 66% as large as
Dnmt1chip/+ mice. Females showed
the same runt phenotype. The error bar indicates ±1 SD,
p < 0.0001 (Student t test, StatView 5.0.1
software). Also shown are growth curves of
Dnmt1chip/chip,
Dnmt1chip/–, and wild-type male mice. Six to
10 mice of each genotype were used for each data point.
(B) Cumulative survival of
Dnmt1chip/– mice. Most
Dnmt1chip/– mice became terminally ill
between 4and 8 months of age. Control mice were
Dnmt1chip/chip (n = 18); experimental
mice were Dnmt1chip/– (n = 33). Mice
were autopsied when visibly ill. At autopsy, 23 of 33
Dnmt1chip/– mice had developed tumors (21
lymphomas and 2 fibrosarcomas). Autopsy of
Dnmt1chip/chip mice at 6 months
(n = 12) and 12 months (n = 6) showed no
evidence of tumor formation. (C) Dß1-to-Jß1
rearrangement at the TCRß locus was analyzed by the polymerase
chain reaction as described (35),
using primers 1 and 4therein. The asterisk denotes the
germline configuration [2171 base pairs (bp)]. When
rearranged, five different amplified fragments are possible,
ranging from 1561 to 381 bp (see wild-type thymus, lanes 1 to
3). Controls also include wild-type tail DNA (lane 14) and
thymus DNA from a recombination-deficient RAG1–/–
mouse. (D) FACS analysis of wild-type thymus (left) or
Dnmt1chip/– tumors (middle and right)
stained for CD4and CD8 receptors, T cell–specific markers.
Tumors analyzed (n = 16) contained either
double-positive CD4high/CD8high cells
(9/16, middle panel) or CD4low/CD8high
cells (7/16, right panel). [View
Larger Version of this Image (34K GIF file)] Fig. 2. Dnmt1
hypomorphs are runted and develop T cell lymphomas. (A)
Average weight of Dnmt1chip/+
(n = 20) and Dnmt1chip/– (n =
20) male littermates at birth (inset).
Dnmt1chip/– mice were 66% as large as
Dnmt1chip/+ mice. Females showed
the same runt phenotype. The error bar indicates ±1 SD,
p < 0.0001 (Student t test, StatView 5.0.1
software). Also shown are growth curves of
Dnmt1chip/chip,
Dnmt1chip/–, and wild-type male mice. Six to
10 mice of each genotype were used for each data point.
(B) Cumulative survival of
Dnmt1chip/– mice. Most
Dnmt1chip/– mice became terminally ill
between 4and 8 months of age. Control mice were
Dnmt1chip/chip (n = 18); experimental
mice were Dnmt1chip/– (n = 33). Mice
were autopsied when visibly ill. At autopsy, 23 of 33
Dnmt1chip/– mice had developed tumors (21
lymphomas and 2 fibrosarcomas). Autopsy of
Dnmt1chip/chip mice at 6 months
(n = 12) and 12 months (n = 6) showed no
evidence of tumor formation. (C) Dß1-to-Jß1
rearrangement at the TCRß locus was analyzed by the polymerase
chain reaction as described (35),
using primers 1 and 4therein. The asterisk denotes the
germline configuration [2171 base pairs (bp)]. When
rearranged, five different amplified fragments are possible,
ranging from 1561 to 381 bp (see wild-type thymus, lanes 1 to
3). Controls also include wild-type tail DNA (lane 14) and
thymus DNA from a recombination-deficient RAG1–/–
mouse. (D) FACS analysis of wild-type thymus (left) or
Dnmt1chip/– tumors (middle and right)
stained for CD4and CD8 receptors, T cell–specific markers.
Tumors analyzed (n = 16) contained either
double-positive CD4high/CD8high cells
(9/16, middle panel) or CD4low/CD8high
cells (7/16, right panel). [View
Larger Version of this Image (34K GIF file)]
|
In addition to the runted phenotype, 80% of
Dnmt1chip/– mice developed aggressive
thymic tumors at 4 to 8 months of age. Cumulative
survival of the Dnmt1chip/– mice is shown
in Fig.
2B. Histological analysis classified the tumors as T
cell lymphomas (11),
and fluorescence-activated cell sorting (FACS) analysis
revealed that most tumors were CD4–/CD8+
or CD4+/CD8+ (Fig.
2D). When tested for D-to-J rearrangements in the T
cell receptor ß locus, four of 10 tumors showed a
predominant Dß1-to-Jß1 rearranged band (Fig.
2C, lanes 5, 9, 11, and 13) consistent with monoclonality.
Tumors without Dß1-to-Jß1 recombination may have
rearranged other D and J elements. Monoclonality suggests
that hypomethylation induces cancer in a precursor cell,
with subsequent events leading to malignant tumor
formation. Consistent with frequent activation of the
c-myc oncogene in mouse and human lymphoma (12),
we found that c-myc was overexpressed in almost
all hypomethylated tumors (15/18 Dnmt1chip/–, Fig.
3C).
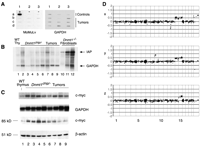 Fig. 3. Expression
and chromosomal analysis of hypomethylated tumors. (A)
RNA slot blot of Dnmt1chip/– lymphomas
(lanes b, c, and d) probed with MMLV cDNA. Also shown are a
positive control lymphoma from a Mov-1 mouse [slot a1 (15)]
and negative control thymuses from wild-type 129/Sv (slot a2)
and a wild-type littermate of a tumor-bearing mouse (slot a3).
(B) Northern blot of IAPs in
Dnmt1chip/– tumors. IAPs can be detected in
most tumors, whereas wild-type thymus does not show IAP
expression. Positive control (lanes 10 to 12, 1:3 serial
dilutions) are Dnmt1–/– hypomethylated
fibroblasts that have been shown to activate IAP expression
(16).
Comparison of IAP and glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) levels shows that most clones express much less IAPs
than the positive control. (C) Expression levels of
c-myc were assessed by Northern blot (top two panels)
and by immunoblot (bottom two panels). Lanes 2 to 7 are tumors
that showed chromosome 15 trisomy; lanes 8 and 9 are tumors
that are diploid for chromosome 15. Probes used were
c-myc exon 2 for the Northern analysis and a rabbit
polyclonal IgG antibody to c-myc for immunoblots (Santa
Cruz Biotechnology). (D) Array comparative genome
hybridization (CGH) analyses of three
Dnmt1chip/– tumors, showing clear
single-copy, whole-chromosome gain of chromosome 15 (x, y, and
z), whole-chromosome gains of 14and loss on distal 12 (x), and
gains of chromosome 14and proximal 9 (y). The X gain (x)
reflects a sex difference between tumor and control. Array CGH
was performed as in (26).
Fluorescence ratios (average of quadruplicate measurements)
for each bacterial artificial chromosome are plotted as a
function of genome location based on the February 2002 freeze
of the assembled mouse genome sequence (http://genome.ucsc.edu/).
Vertical lines delimit chromosome boundaries. [View
Larger Version of this Image (47K GIF file)] Fig. 3. Expression
and chromosomal analysis of hypomethylated tumors. (A)
RNA slot blot of Dnmt1chip/– lymphomas
(lanes b, c, and d) probed with MMLV cDNA. Also shown are a
positive control lymphoma from a Mov-1 mouse [slot a1 (15)]
and negative control thymuses from wild-type 129/Sv (slot a2)
and a wild-type littermate of a tumor-bearing mouse (slot a3).
(B) Northern blot of IAPs in
Dnmt1chip/– tumors. IAPs can be detected in
most tumors, whereas wild-type thymus does not show IAP
expression. Positive control (lanes 10 to 12, 1:3 serial
dilutions) are Dnmt1–/– hypomethylated
fibroblasts that have been shown to activate IAP expression
(16).
Comparison of IAP and glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) levels shows that most clones express much less IAPs
than the positive control. (C) Expression levels of
c-myc were assessed by Northern blot (top two panels)
and by immunoblot (bottom two panels). Lanes 2 to 7 are tumors
that showed chromosome 15 trisomy; lanes 8 and 9 are tumors
that are diploid for chromosome 15. Probes used were
c-myc exon 2 for the Northern analysis and a rabbit
polyclonal IgG antibody to c-myc for immunoblots (Santa
Cruz Biotechnology). (D) Array comparative genome
hybridization (CGH) analyses of three
Dnmt1chip/– tumors, showing clear
single-copy, whole-chromosome gain of chromosome 15 (x, y, and
z), whole-chromosome gains of 14and loss on distal 12 (x), and
gains of chromosome 14and proximal 9 (y). The X gain (x)
reflects a sex difference between tumor and control. Array CGH
was performed as in (26).
Fluorescence ratios (average of quadruplicate measurements)
for each bacterial artificial chromosome are plotted as a
function of genome location based on the February 2002 freeze
of the assembled mouse genome sequence (http://genome.ucsc.edu/).
Vertical lines delimit chromosome boundaries. [View
Larger Version of this Image (47K GIF file)]
|
Genomic hypomethylation may contribute to lymphomagenesis by
an epigenetic or a genetic mechanism. We considered three
possible mechanisms.
- Hypomethylation may induce endogenous retroviral elements,
leading in turn to insertional activation of
proto-oncogenes (13).
To test this idea, we hybridized RNA from randomly
selected tumors with a Moloney murine leukemia virus
(MMLV) cDNA probe and an IAP probe to detect endogenous
retroviral and IAP expression, respectively. Of nine
Dnmt1chip/– tumors, none showed
C-type retroviral activation (Fig.
3A) (14)
and only one of eight tumors showed a moderate increase
in IAP expression (Fig.
3B, lane 7). In contrast, strong C-type retroviral
expression was seen in a MMLV-induced lymphoma [Fig.
3A, slot a1 (15)]
and IAP expression was highly activated in
Dnmt1–/– fibroblasts [Fig.
3B, lanes 10 to 12 (16)].
Because c-myc is a frequent target for
insertional activation by retroviral elements (17),
we searched for inserted proviral elements in hypomethylated
and MMLV-induced tumors. In 3 of 12 MMLV-induced tumors,
an insertional rearrangement was seen in the vicinity
of the c-myc locus, in agreement with previous
observations (17).
In contrast, no rearrangements were detected in
hypomethylated tumors [0/18 (11)].
We conclude that the extent of hypomethylation in
Dnmt1chip/– mice does not effectively
activate endogenous retroviral elements and that virus
insertions may not be a prevalent mechanism in
hypomethylation-induced lymphoma.
- Hypomethylation may activate protooncogenes through
epigenetic effects (18,
19).
Indeed, c-myc was overexpressed in most
hypomethylated tumors (Fig.
3C). However, it is unlikely that activation
of c-myc is a direct consequence of
promoter demethylation because the gene is
expressed at normal levels in thymuses from 2- and
4-week-old mice that show a level of
hypomethylation identical to that of the tumors
[Fig.
1D (11)].
In addition, c-myc was not activated in
Dnmt–/– fibroblasts that are
almost completely demethylated (16).
Finally, if oncogene activation by
hypomethylation stimulated T cell proliferation as a
first step in transformation, one would expect
the lymphomas to be polyclonal rather than
monoclonal (Fig.
2C).
- Hypomethylation may induce genomic instability. In fact,
a significantly increased frequency of
chromosomal rearrangements such as loss of
heterozygosity (LOH) was observed in Dnmt1 mutant
ES cells, suggesting that normal levels of
methylation are important for genomic
stability (20).
Defects in DNA methylation have been linked
to genome instability in studies of colorectal tumor
cell lines (21),
mouse tumor models (22,
23),
and patients with immunodeficiency–centromeric
instability–facial anomalies (ICF) syndrome (24,
25).
To test whether DNA hypomethylation increases genomic
instability in Dnmt1chip/– tumors, we
performed array-based comparative genome hybridization
[array CGH (26)]
using thymic tumor genomic DNA prepared from
Dnmt1chip/– and Mov-1 (15)
and Mov-14 (27)
MMLV transgenic mice (Fig.
3D). There was a statistically significant difference
in chromosome gains between these tumor classes (Table
1). Ten of 12 hypomethylated tumors exhibited a gain
of chromosome 15, whereas only 2 of 12 MMLV-induced tumors
showed this change (P = 0.004). Relative to
MMLV-induced tumors, hypomethylated tumors also displayed
a gain of chromosome 14 (4/12 versus 0/12, P =
0.05) and a higher degree of duplicated and deleted
chromosome regions (Table
1) (Fig.
3D).
Table 1. Gains or losses of chromosomes
in Dnmt1chip/- and MMLV-induced tumors.
The numbers indicate the number of times a particular event
occurred in the Dnmt1chip/- or Moloney
tumors. These events were not mutually exclusive; many
tumors exhibited multiple chromosomal events.
| Chromosomal changes
|
Dnmt1chip/-
tumors (n = 12) |
MMLV-induced tumors
(n = 12) |
|
| Chr 15 gain
|
10
|
2
|
| Chr 14 gain
|
4
|
0
|
| Chr 10 gain
|
0
|
1
|
| Partial Chr 9 gain
|
2
|
0
|
| Partial Chr 4 gain
|
1
|
0
|
| Partial Chr 16 loss
|
1
|
0
|
Partial Chr 12 loss
|
1
|
0
| |
| |
Together with the centromeric hypomethylation we observed (Fig.
1C), these results suggest a causal link between DNA
hypomethylation and chromosomal instability as one
mechanism leading to tumorigenesis. The increased
fluorescence ratios observed for chromosomes 14 and 15
are consistent with singlecopy whole-chromosome gains
throughout the tumor (Fig.
3D), which suggests that they are early events in the
development of these monoclonal T cell lymphomas.
Chromosome 15 is frequently duplicated in mouse T cell
tumors (28,
29)
and contains the oncogene c-myc, which when
overexpressed causes T cell lymphomas (17).
The fact that c-myc is overexpressed (RNA and
protein) in most hypomethylated tumors (Fig.
3C) is consistent with a mechanism in which a gain of
chromosome 15 contributes, at least in part, to the
elevated expression of c-myc. Moreover,
c-myc expression was lower in the two tumors that
did not show trisomy 15 than in the other tumors (Fig.
3C).
Our results show that genomic hypomethylation causes
tumorigenesis in mice and is associated with the
acquisition of additional genomic changes. Consistent
with this, genomic hypomethylation was found to promote
tumorigenesis in a different mouse tumor model and to
increase the rate of LOH in cultured fibroblasts (23).
However, it remains possible that DNA hypomethylation
contributes to tumorigenesis through other mechanisms
unrelated to chromosomal instability. The phenotype of
hypomethylated mice is also consistent with that of
Suv39h histone methyltransferase mutant mice;
hence, DNA and histone methylation, pericentric chromatin
structure, and the maintenance of chromosomal stability
may be linked (30).
DNA methyltransferase inhibitors such as
5-aza-2'-deoxycytidine have been used successfully to
treat cancer in humans (19,
31)
and mice (32,
33).
The efficacy of these drugs is presumably due to their
ability to reverse the epigenetic silencing of tumor
suppressor genes. In light of our results, however, this
therapeutic strategy should perhaps be considered a
double-edged sword: Genomic demethylation may protect
against some cancers such as intestinal tumors in the
ApcMin mouse model (32)
but may promote genomic instability and LOH (20,
23)
and increase the risk of cancer in other tissues, as seen
in hypomethylated mutant mice.
References and
Notes
| 1. |
P. A. Jones, S. B. Baylin, Nature
Rev. Genet. 3, 415 (2002).[ISI][Medline] |
| 2. |
A. P. Feinberg, B. Vogelstein,
Nature 301, 89 (1983).[ISI][Medline] |
| 3. |
M. Ehrlich, Oncogene
21, 5400 (2002).[CrossRef][ISI][Medline] |
| 4. |
M. A. Gama-Sosa, Nucleic Acids
Res. 11, 6883 (1983).[Abstract] |
| 5. |
J. N. Lapeyre, F. F. Becker,
Biochem. Biophys. Res. Commun. 87, 698 (1979).[ISI][Medline] |
| 6. |
R. Jaenisch, A. Bird, Nature
Genet. 33 (suppl.), 245 (2003).[CrossRef][ISI][Medline] |
| 7. |
H. Lei et al.,
Development 122, 3195 (1996).[Abstract/Free Full Text] |
| 8. |
E. Li, T. H. Bestor, R. Jaenisch,
Cell 69, 915 (1992).[ISI][Medline] |
| 9. |
K. L. Tucker et al., Genes
Dev. 10, 1008 (1996).[Abstract] |
| 10. |
C. P. Walsh, J. R. Chaillet, T. H.
Bestor, Nature Genet. 20, 116 (1998).[CrossRef][ISI][Medline] |
| 11. |
F. Gaudet, A. Eden, R. Jaenisch,
unpublished data. |
| 12. |
S. Cory, D. L. Vaux, A. Strasser, A.
W. Harris, J. M. Adams, Cancer Res. 59 (suppl.),
1685s (1999).[ISI][Medline] |
| 13. |
R. Jaenisch, A. Schnieke, K.
Harbers, Proc. Natl. Acad. Sci. U.S.A. 82, 1451
(1985).[ISI][Medline] |
| 14. |
R. Jaenisch, Proc. Natl. Acad.
Sci. U.S.A. 73, 1260 (1976).[ISI][Medline] |
| 15. |
D. Jahner, R. Jaenisch,
Nature 287, 456 (1980).[ISI][Medline] |
| 16. |
L. Jackson-Grusby et al.,
Nature Genet. 27, 31 (2001).[CrossRef][ISI][Medline] |
| 17. |
G. Selten, H. T. Cuypers, M.
Zijlstra, C. Melief, A. Berns, EMBO J. 3, 3215
(1984).[Abstract] |
| 18. |
E. Wainfan, L. A. Poirier, Cancer
Res. 52, 2071s (1992).[Abstract] |
| 19. |
A. R. Karpf, D. A. Jones,
Oncogene 21, 5496 (2002).[CrossRef][ISI][Medline] |
| 20. |
R. Z. Chen, U. Pettersson, C. Beard,
L. Jackson-Grusby, R. Jaenisch, Nature 395, 89
(1998).[CrossRef][ISI][Medline] |
| 21. |
C. Lengauer, K. W. Kinzler, B.
Vogelstein, Proc. Natl. Acad. Sci. U.S.A. 94,
2545 (1997).[Abstract/Free Full Text] |
| 22. |
B. N. Trinh, T. I. Long, A. E.
Nickel, D. Shibata, P. W. Laird, Mol. Cell. Biol.
22, 2906
(2002).[Abstract/Free Full Text] |
| 23. |
A. Eden, F. Gaudet, A. Waghmare, R.
Jaenisch, Science 300, 455 (2003).[Free Full Text] |
| 24. |
M. Jeanpierre et al., Hum.
Mol. Genet. 2, 731 (1993).[Abstract] |
| 25. |
G. L. Xu et al.,
Nature 402, 187 (1999).[CrossRef][ISI][Medline] |
| 26. |
G. Hodgson et al., Nature
Genet. 29, 459 (2001).[CrossRef][ISI][Medline] |
| 27. |
C. Stewart, K. Harbers, D. Jahner,
R. Jaenisch, Science 221, 760 (1983).[ISI][Medline] |
| 28. |
Z. Wirschubsky, P. Tsichlis, G.
Klein, J. Sumegi, Int. J. Cancer 38, 739 (1986).[ISI][Medline] |
| 29. |
M. Muto, Y. Chen, E. Kubo, K. Mita,
Jpn. J. Cancer Res. 87, 247 (1996).[ISI][Medline] |
| 30. |
A. H. F. M. Peters et al.,
Cell 107, 323 (2001).[ISI][Medline] |
| 31. |
V. Zagonel et al.,
Leukemia 7 (suppl. 1), 30 (1993).[ISI][Medline] |
| 32. |
P. W. Laird et al.,
Cell 81, 197 (1995).[ISI][Medline] |
| 33. |
A. R. MacLeod, M. Szyf, J. Biol.
Chem. 270, 8037 (1995).[Abstract/Free Full Text] |
| 34. |
F. Gaudet, D. Talbot, H. Leonhardt,
R. Jaenisch, J. Biol. Chem. 273, 32725 (1998).[Abstract/Free Full Text] |
| 35. |
C. E. Whitehurst, S. Chattopadhyay,
J. Chen, Immunity 10, 313 (1999).[ISI][Medline] |
| 36. |
We thank R. Flannery for help with
the mouse colony, and K. Hong and C. Cardoso for helpful
discussions. Supported by grants from the Max Delbrück Center
and the Deutsche Forschungsgemeinschaft (H.L.), by NIH grant
CA87869 (R.J.), and by EMBO fellowship ALTF 43-1999 and
Boehringer Ingelheim (A.E.). | 18
February 2003; accepted 10 March
2003
10.1126/science.1083558
Include this information when
citing this paper.
This article has been cited by other
articles:
- Cordier, S., Monfort, C., Filippini, G., Preston-Martin, S.,
Lubin, F., Mueller, B. A., Holly, E. A., Peris-Bonet, R.,
McCredie, M., Choi, W., Little, J., Arslan, A. (2004). Parental
Exposure to Polycyclic Aromatic Hydrocarbons and the Risk of
Childhood Brain Tumors: The SEARCH International Childhood Brain
Tumor Study. Am. J. Epidemiol. 159: 1109-1116 [Abstract]
[Full
Text]
- De Smet, C., Loriot, A., Boon, T. (2004). Promoter-Dependent
Mechanism Leading to Selective Hypomethylation within the 5'
Region of Gene MAGE-A1 in Tumor Cells. Mol. Cell. Biol.
24: 4781-4790 [Abstract]
[Full
Text]
- Sun, L.-Q., Lee, D. W., Zhang, Q., Xiao, W., Raabe, E. H.,
Meeker, A., Miao, D., Huso, D. L., Arceci, R. J. (2004). Growth
retardation and premature aging phenotypes in mice with disruption
of the SNF2-like gene, PASG. Genes & Dev. 18:
1035-1046 [Abstract]
[Full
Text]
- Komarova, N. L., Wodarz, D. (2004). The optimal rate of
chromosome loss for the inactivation of tumor suppressor genes in
cancer. Proc. Natl. Acad. Sci. U. S. A. 101: 7017-7021
[Abstract]
[Full
Text]
- Yanagawa, N., Tamura, G., Honda, T., Endoh, M., Nishizuka, S.,
Motoyama, T. (2004). Demethylation of the Synuclein {gamma} Gene
CpG Island in Primary Gastric Cancers and Gastric Cancer Cell
Lines. Clin Cancer Res 10: 2447-2451 [Abstract]
[Full
Text]
- Wada, K., Maesawa, C., Akasaka, T., Masuda, T. (2004).
Aberrant Expression of the Maspin Gene Associated with Epigenetic
Modification in Melanoma Cells. J Invest Dermatol 122:
805-811 [Abstract]
[Full
Text]
- Liu, Z., Fisher, R. A. (2004). RGS6 Interacts with DMAP1 and
DNMT1 and Inhibits DMAP1 Transcriptional Repressor Activity.
J. Biol. Chem. 279: 14120-14128 [Abstract]
[Full
Text]
- Yoshida, M., Nosaka, K., Yasunaga, J.-i., Nishikata, I.,
Morishita, K., Matsuoka, M. (2004). Aberrant expression of the
MEL1S gene identified in association with hypomethylation in adult
T-cell leukemia cells. Blood 103: 2753-2760 [Abstract]
[Full
Text]
- Dodge, J. E., Kang, Y.-K., Beppu, H., Lei, H., Li, E. (2004).
Histone H3-K9 Methyltransferase ESET Is Essential for Early
Development. Mol. Cell. Biol. 24: 2478-2486 [Abstract]
[Full
Text]
- Xin, H., Yoon, H.-G., Singh, P. B., Wong, J., Qin, J. (2004).
Components of a Pathway Maintaining Histone Modification and
Heterochromatin Protein 1 Binding at the Pericentric
Heterochromatin in Mammalian Cells. J. Biol. Chem. 279:
9539-9546 [Abstract]
[Full
Text]
- Ghoshal, K., Majumder, S., Datta, J., Motiwala, T., Bai, S.,
Sharma, S. M., Frankel, W., Jacob, S. T. (2004). Role of Human
Ribosomal RNA (rRNA) Promoter Methylation and of
Methyl-CpG-binding Protein MBD2 in the Suppression of rRNA Gene
Expression. J. Biol. Chem. 279: 6783-6793 [Abstract]
[Full
Text]
- Gaudet, F., Rideout, W. M. III, Meissner, A., Dausman, J.,
Leonhardt, H., Jaenisch, R. (2004). Dnmt1 Expression in Pre- and
Postimplantation Embryogenesis and the Maintenance of IAP
Silencing. Mol. Cell. Biol. 24: 1640-1648 [Abstract]
[Full
Text]
- Karpf, A. R., Lasek, A. W., Ririe, T. O., Hanks, A. N.,
Grossman, D., Jones, D. A. (2004). Limited Gene Activation in
Tumor and Normal Epithelial Cells Treated with the DNA
Methyltransferase Inhibitor 5-Aza-2'-deoxycytidine. Mol
Pharmacol 65: 18-27 [Abstract]
[Full
Text]
- Silva, S., Kovalchuk, A. L., Kim, J. S., Klein, G., Janz, S.
(2003). BCL2 Accelerates Inflammation-induced BALB/c Plasmacytomas
and Promotes Novel Tumors with Coexisting T(12;15) and T(6;15)
Translocations. Cancer Res 63: 8656-8663 [Abstract]
[Full
Text]
- Rabinowicz, P. D., Palmer, L. E., May, B. P., Hemann, M. T.,
Lowe, S. W., McCombie, W. R., Martienssen, R. A. (2003). Genes and
Transposons Are Differentially Methylated in Plants, but Not in
Mammals. Genome Res. 13: 2658-2664 [Abstract]
[Full
Text]
- Kopelovich, L., Crowell, J. A., Fay, J. R. (2003). The
Epigenome as a Target for Cancer Chemoprevention. J Natl
Cancer Inst 95: 1747-1757 [Abstract]
[Full
Text]
- Fang, M. Z., Wang, Y., Ai, N., Hou, Z., Sun, Y., Lu, H.,
Welsh, W., Yang, C. S. (2003). Tea Polyphenol
(-)-Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and
Reactivates Methylation-Silenced Genes in Cancer Cell Lines.
Cancer Res 63: 7563-7570 [Abstract]
[Full
Text]
- Herman, J. G., Baylin, S. B. (2003). Gene Silencing in Cancer
in Association with Promoter Hypermethylation. N Engl J
Med 349: 2042-2054 [Full
Text]
- Eden, A., Gaudet, F., Jaenisch, R. (2003). Response to Comment
on "Chromosomal Instability and Tumors Promoted by DNA
Hypomethylation" and "Induction of Tumors in Mice by Genomic
Hypomethylation". Science 302: 1153c-1153 [Full
Text]
- Yang, A. S., Estecio, M. R.H., Garcia-Manero, G., Kantarjian,
H. M., Issa, J.-P. J. (2003). Comment on "Chromosomal Instability
and Tumors Promoted by DNA Hypomethylation" and "Induction of
Tumors in Mice by Genomic Hypomethylation". Science 302:
1153b-1153 [Full
Text]
- Hiltunen, M. O., Yla-Herttuala, S. (2003). DNA Methylation,
Smooth Muscle Cells, and Atherogenesis. Arterioscler Thromb
Vasc Biol 23: 1750-1753 [Abstract]
[Full
Text]
Related articles in Science:
- Chromosomal Instability and Tumors Promoted by DNA
Hypomethylation
- Amir Eden, François Gaudet, Alpana Waghmare, and Rudolf
Jaenisch
Science 2003 300: 455. (in Brevia) [Full
Text]
- CANCER:
An Unstable
Liaison
- Christoph Lengauer
Science 2003 300: 442-443. (in
Perspectives) [Summary]
[Full
Text]
- Comment on "Chromosomal Instability and Tumors
Promoted by DNA Hypomethylation" and "Induction of Tumors in Mice
by Genomic Hypomethylation"
- Allen S. Yang, Marcos R.H. Estecio, Guillermo Garcia-Manero,
Hagop M. Kantarjian, and Jean-Pierre J. Issa
Science 2003 302:
1153. (in Technical Comments) [Full
Text]
- Response to Comment on "Chromosomal Instability and
Tumors Promoted by DNA Hypomethylation" and "Induction of Tumors
in Mice by Genomic Hypomethylation"
- Amir Eden, François Gaudet, and Rudolf Jaenisch
Science
2003 302: 1153. (in Technical Comments) [Full
Text]
Volume 300, Number 5618, Issue of 18 Apr 2003, pp. 489-492.
Copyright © 2003 by The American Association for the
Advancement of Science. All rights reserved.
|




