
Science, Vol 300, Issue 5618,
442-443 , 18 April 2003
[DOI: 10.1126/science.1084468]
CANCER:
An Unstable
LiaisonChristoph Lengauer*
Changes in the pattern of DNA methylation are
common in human tumors (1).
Both genome-wide hypomethylation and region-specific
hypermethylation seem to be important in the formation and
progression of cancers (carcinogenesis). The global level of DNA
methylation is generally lower in tumor cells than in normal cells
(2),
but this hypomethylation is curious in light of the increased
expression of DNA methyltransferase (an enzyme that adds methyl
groups to specific cytosines in DNA) in many tumor cells (3).
In two papers on pages 455
and 489
of this issue (4,
5),
Jaenisch's group presents evidence for a potential link between DNA
hypomethylation, genomic instability, and cancer.
DNA hypermethylation is associated with the inappropriate
transcriptional silencing of tumor suppressor genes, explaining its
pervasive role in oncogenesis. The biological significance of DNA
hypomethylation in cancer is less clear. Early experiments using DNA
methylation inhibitors in vivo and in vitro seemed to support the
involvement of DNA hypomethylation in carcinogenesis. Such
experiments resulted in conversion of low-metastatic tumor cell
lines to high-metastatic versions and formation of transformed foci.
Feeding methyl-deficient diets to rats and mice resulted in global
DNA undermethylation, the formation of liver tumors, and
demethylation of proto-oncogenes (6).
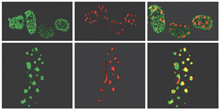
DNMT1 and DNA methylation.
(Top) In cultured mouse
cells in early S phase of the cell cycle, DNMT1 (green) is
distributed into small foci where it maintains the sparse
methylation pattern of early-replicating euchromatic regions of the
DNA (red). (Bottom) During late S-phase
replication, DNMT1 (green) becomes localized to densely methylated
blocks of heterochromatin (red) where it maintains the
hypermethylation of these silenced regions. (Top
and Bottom) The images on the far right represent
the superimposition of the first two images; regions of overlap
appear in yellow.
CREDIT: K. E. BACHMAN/SIDNEY KIMMEL COMPREHENSIVE
CANCER CENTER, JOHNS HOPKINS UNIVERSITY
Two analyses of DNA methyltransferase-deficient mice have
complicated the interpretation of established hypotheses about
methylation and cancer. Jaenisch's group reduced DNA
methyltransferase 1 (Dnmt1) activity in the Min mouse model, which
mimics familial adenomatous polyposis, a human disease characterized
by formation of numerous precancerous polyps in the colon. They
achieved this by giving a low dose of 5-azadeoxycytidine to mice
heterozygous for the DNA methyltransferase gene. The decrease in DNA
methylation significantly reduced the number of intestinal adenomas
formed in the mice (7).
These results conflicted with conventional wisdom at the time
because they implied that DNA hypomethylation had no oncogenic
effect. Further confusing the issue, Trinh et al. found
that a reduction in DNMT1 activity had significant but opposing
effects on the development of two different types of tumor (8).
To study the interaction between DNA mismatch repair deficiency and
DNA methylation, they introduced Dnmt1 mutations into a mouse strain
deficient in the MLH1 protein, which is required for repair of
mistakes made in DNA replication (8).
Mice harboring hypomorphic Dnmt1 mutations showed diminished DNA
methylation but developed normally and were tumor-free. When crossed
with Mlh1-/- homozygous mice lacking the MLH1 protein,
the resulting offspring were less likely to develop the intestinal
cancers characteristic of their mismatch repair-deficient parent.
However, these same mice developed invasive T and B cell lymphomas
earlier and at a much higher frequency than did their Dnmt1
wild-type littermates.
How does hypomethylation influence neoplasia? Because both
genomic instability and hypomethylation are observed early during
carcinogenesis (1,
9),
it is tempting to speculate that genomic hypomethylation, by
destabilizing the genome, provides the incipient cancer cells with a
way of acquiring more mutations. Earlier studies have suggested that
defects in DNA methylation might contribute to the chromosomal
instability observed in aneuploid human colorectal cancer cell lines
(10).
Also, DNA hypomethylation of very discrete locations within the
genome has been associated with abnormal chromosomal structures,
such as those observed in cells from patients with ICF syndrome
(immunodeficiency-centromeric instability- facial abnormalities).
This rare recessive disease is caused by mutations in the catalytic
domain of the Dnmt3b gene, which encodes another DNA
methyltransferase (11).
Murine embryonic stem (ES) cells lacking Dnmt1 exhibit an increased
frequency of chromosomal deletions (12).
In their two new studies, Jaenisch's group now extends its ES cell
work to somatic cells (4,
5).
Mice carrying two Dnmt1 null alleles die during
gestation (13).
To overcome this problem, Jaenisch and colleagues (5)
combined a hypomorphic allele (Dnmt1chip) with a
null allele to generate Dnmt1chip/- compound
heterozygote animals with substantially reduced DNA methylation (10%
of wild type). Such a genetic approach is superior to previous
pharmacological studies by the same group (7)
because the Dnmt1 hypomorphic allele causes genome-wide
hypomethylation in all tissues while avoiding the detrimental
effects of mutations. This severe DNA hypomethylation is sufficient
to induce formation of T cell lymphomas. Using array-based
comparative genomic hybridization, the authors compared genomic DNA
from Dnmt1chip/- tumors with that from Moloney
virus-induced tumors. They observed a subtle but statistically
significant increase in gains and losses of chromosomes in the
hypomethylated tumors (5).
In their companion study, the investigators provide further
support for the potential destabilizing effect of hypomethylation on
genomic DNA (4).
Purposely avoiding models in which hypermethylation and gene
silencing might be involved, they selected a different mouse model
in which a chromosomal event was the rate-limiting step and
essential for tumor formation (4).
They introduced a hypomorphic Dnmt1chip/- allele
into animals with mutations in p53 and Nf1 alleles
on the same copy of chromosome 11. Loss of Nf1 activates the
proto-oncogene ras and cooperates with inactivating
mutations in the p53 tumor suppressor gene during malignant
transformation. All of the mice harboring null Nf1 and
p53 alleles developed soft tissue sarcomas between 3 and 7
months of age (14).
These sarcomas exhibited loss of heterozygosity (LOH) at both gene
loci. Consistent with the fact that DNA hypomethylation increased
the rate of LOH in cultured fibroblasts, sarcomas formed earlier in
the hypomethylated animals (4).
This phenotype is strikingly consistent with that of histone
methyltransferase (Suv39h) mutant mice, which provides a compelling
link between DNA and histone methylation, pericentric chromatin
structure, and the maintenance of chromosomal stability (15).
The new data provide the most direct evidence so far for an
effect of hypomethylation on chromosomal stability. However, the
mechanisms by which hypomethylation causes genomic instability
remain unclear. The authors show that in hypomethylated cells, LOH
occurs preferentially in the centromeric region, but they have been
unable to determine whether loss of the region carrying the
wild-type copies of Nf1 and p53 is the result of
mitotic recombination or of whole chromosome loss. Identifying cells
with only one copy of chromosome 11 (per diploid genome) would be an
indication of whole chromosome loss, but such an analysis is
complicated by the fact that both methylated and hypomethylated
tumor cells are mostly aneuploid (16).
Are these mouse data relevant to human cancers? Maybe. Although
DNMT1 accounts for most of the methylation in normal mouse cells,
human colorectal cancer cells lacking Dnmt1 retain significant
genomic methylation and associated gene silencing (17).
Rhee et al. have also disrupted the Dnmt3b gene in
human cell lines that codes for another DNA methyltransferase (18).
This deletion reduced global DNA methylation by less than 3%.
Surprisingly, genetic disruption of both Dnmt1 and
Dnmt3b in human cell lines nearly eliminated
methyltransferase activity and reduced genomic DNA methylation by
more than 95%. These marked changes resulted in demethylation of
repeated sequences, loss of insulin-like growth factor II
imprinting, abrogation of silencing of the tumor suppressor gene
p16INK4a, and growth suppression (18).
These results provide compelling evidence that the two enzymes
cooperatively maintain DNA methylation and gene silencing in human
colorectal cancer, and that such methylation is essential for
optimal neoplastic proliferation. However, disruption of
Dnmt1 and/or Dnmt3b did not lead to a dramatically
increased rate of gain or loss of chromosomes in these cells (18).
Again, such discrepancies might reflect differences in model
systems, varying mechanisms in different species, or tissue
specificity. Therefore, any implications for the treatment of human
cancers need to be drawn with extreme caution. DNA methyltransferase
inhibitors such as 5-aza-CdR have shown some efficacy in treating
leukemias (19).
DNA hypomethylation seems to promote LOH and genomic instability,
and so DNA methyltransferase inhibitors might fatally accelerate
tumor progression by increasing chromosomal instability just enough
to promote tumorigenesis. Alternatively, DNA methyltransferase
inhibitors could drive the cancer to self- destruct by increasing
chromosomal instability enough to push tumor cells with already
unstable genomes into death (20).
This model explains why genomic demethylation may protect against
some cancers such as intestinal tumors in the APCMin
mouse model (7),
but may increase the risk of cancers in other tissues, exemplified
by the tumors arising in the hypomethylated mutant mice.
References and Notes
- P. A. Jones, S. B. Baylin, Nature Rev. Genet.
3, 415 (2002) [Medline].
- A. P. Feinberg, B. Vogelstein, Nature
301, 89 (1983) [Medline].
- T. L. Kautiainen, P. A. Jones, J. Biol. Chem.
261, 1594 (1986) [Medline].
- A. Eden et al., Science
300, 455
(2003).
- F. Gaudet et al., Science
300, 489
(2003).
- M. Ehrlich, Oncogene 21, 5400 (2002)
[Medline].
- P. Laird et al., Cell 81,
197 (1995) [Medline].
- B. N. Trinh et al., Mol. Cell. Biol.
22, 2906 (2002) [Medline].
- C. Lengauer et al., Nature
396, 643 (1998) [Medline].
- ------, Proc. Natl. Acad. Sci. U.S.A.
94, 2545 (1997) [Medline].
- G.-L. Xu et al., Nature
402, 187 (1999) [Medline].
- R. Z. Chen et al., Nature
395, 89 (1998) [Medline].
- E. Li et al., Cell 69, 915
(1992) [Medline].
- K. S. Vogel et al., Science
286, 2176
(1999).
- A. H. Peters et al., Cell
107, 323 (2001) [Medline].
- R. Jaenisch, personal communication.
- I. Rhee et al., Nature 404,
1003 (2000) [Medline].
- I. Rhee et al., Nature 416,
552 (2002) [Medline].
- A. R. Karpf, D. A. Jones, Oncogene
21, 5496 (2002) [Medline].
- D. P. Cahill et al., Trends Cell Biol.
9, M57 (1999) [Medline].
- I thank H. Rajagopalan and B. Vogelstein for their helpful
comments.
The author is in the Department of Oncology, Sidney Kimmel
Comprehensive Cancer Center, Johns Hopkins University School of
Medicine, Baltimore, MD 21231, USA. E-mail: lengauer@jhmi.edu
10.1126/science.1084468
Include this information when citing this
paper.
This article has been cited by other
articles:
- Yanagawa, N., Tamura, G., Honda, T., Endoh, M., Nishizuka, S.,
Motoyama, T. (2004). Demethylation of the Synuclein {gamma} Gene
CpG Island in Primary Gastric Cancers and Gastric Cancer Cell
Lines. Clin Cancer Res 10: 2447-2451 [Abstract]
[Full
Text]
- Yang, A. S., Estecio, M. R.H., Garcia-Manero, G., Kantarjian,
H. M., Issa, J.-P. J. (2003). Comment on "Chromosomal Instability
and Tumors Promoted by DNA Hypomethylation" and "Induction of
Tumors in Mice by Genomic Hypomethylation". Science 302:
1153b-1153 [Full
Text]
Related articles in Science:
- Chromosomal Instability and Tumors Promoted by DNA
Hypomethylation
- Amir Eden, François Gaudet, Alpana Waghmare, and Rudolf
Jaenisch
Science 2003 300: 455. (in Brevia) [Full
Text]
- Induction of Tumors in Mice by Genomic
Hypomethylation
- François Gaudet, J. Graeme Hodgson, Amir Eden, Laurie
Jackson-Grusby, Jessica Dausman, Joe W. Gray, Heinrich Leonhardt,
and Rudolf Jaenisch
Science 2003 300: 489-492. (in Reports)
[Abstract]
[Full
Text]
Volume 300, Number 5618, Issue of 18 Apr 2003, pp. 442-443.
Copyright © 2003 by The American Association for the
Advancement of Science. All rights reserved.
|




