 |
 |
Science, Vol 303, Issue 5658, 666-669 , 30 January 2004
Reactivation of the Paternal X Chromosome in Early Mouse EmbryosIt is generally accepted that paternally imprinted X inactivation occurs exclusively in extraembryonic lineages of mouse embryos, whereas cells of the embryo proper, derived from the inner cell mass (ICM), undergo only random X inactivation. Here we show that imprinted X inactivation, in fact, occurs in all cells of early embryos and that the paternal X is then selectively reactivated in cells allocated to the ICM. This contrasts with more differentiated cell types where X inactivation is highly stable and generally irreversible. Our observations illustrate that an important component of genome plasticity in early development is the capacity to reverse heritable gene silencing decisions. 1 X inactivation group, MRC Clinical Sciences Centre,
ICSM, Hammersmith Hospital, London, W12 0NN, UK. * To whom correspondence should be addressed. E-mail: neil.brockdorff@csc.mrc.ac.uk
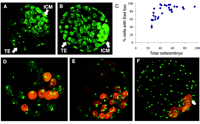
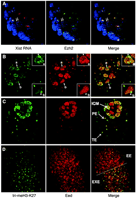
X inactivation is the developmentally regulated silencing of one of the two X chromosomes in female mammals, providing the mechanism for dosage compensation of X-linked genes relative to XY males. In mouse embryos, there is imprinted X inactivation of the paternal X chromosome (Xp) in the extraembryonic trophectoderm (TE) and primitive endoderm (PE). In all other cells, X inactivation is random. Establishment of these patterns has been thought to occur in a lineage-specific manner, with imprinted X inactivation initiated only in TE and PE cells as they differentiate at the blastocyst stage. In contrast, ICM cells, which give rise to the embryo proper, have been thought to retain both X chromosomes in the active state until they differentiate and undergo random X inactivation during early postimplantation development (1–5). Paradoxically, Xist RNA, the cis-acting signal that initiates X inactivation, is expressed from Xp as early as the two-cell stage (6, 7). This has been rationalized by supposing that cells of the early embryo cannot respond to Xist RNA (6). In a recent study, we have shown that recruitment of the Eed-Ezh2 Polycomb-Group (PcG) complex to the inactive X (Xi), is required to establish trimethylation of histone H3 lysine-27 (H3-K27) in postimplantation embryos (8). In the course of analyzing localization of Eed-Ezh2 on Xi in preimplantation embryos, we found that PcG localization occurs in all cells of early- and mid-stage XX blastocysts, including those corresponding morphologically to the ICM (Fig. 1A). In contrast, localization to Xi was no longer detectable in the ICM region of late-stage blastocysts, despite high levels of the Eed-Ezh2 proteins in the nuclei (Fig. 1B). Scoring data demonstrate that only early-stage blastocysts have Eed-Ezh2 foci in 100% of cells (Fig. 1C). In dual staining experiments, Ezh2 was seen to colocalize with Eed at all stages analyzed and Eed-Ezh2 foci colocalized with Xist RNA domains (fig. S1, A to C).
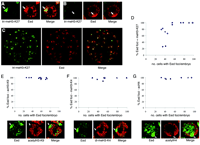
Eed-Ezh2 localization to Xi in the ICM was confirmed using dual staining for Eed and the recently described homeodomain protein Nanog, expressed specifically in ICM cells (9, 10). Eed foci were detectable in all Nanog-positive cells in early- and mid-stage blastocysts (Fig. 1, D and E) but were progressively lost at later stages (Fig. 1F). Interestingly, loss of Eed foci in the ICM region of maturing blastocysts occurs specifically in Nanog-positive cells, representing precursors of the embryo proper, but does not occur in Nanog-negative cells, which represent the primitive endoderm lineage. Our observations led us to consider that imprinted X inactivation may occur in all cells of early blastocysts and may then be selectively reversed in the ICM, establishing the ground state for subsequent random X inactivation. To determine whether early ICM cells exhibit other markers of X inactivation, we carried out dual labeling for Eed and specific modifications on histone N-termini, namely trimethylation of H3-K27, deacetylation of H3-K9, loss of methylation at H3-K4, and deacetylaton of H4 (8, 11–13) (fig. S2, A to D). Eed localization to Xi in XX embryos is first detectable in morula (8). At this stage, H3-K27 methylation on Xi was only detectable in some cells with Eed foci (Fig. 2, A, B, and D). The proportion increased rapidly thereafter, and by blastocyst stage H3-K27 methylation on Xi was detected in the majority of cells, including in the ICM region (Fig. 2, C and D). Hypoacetylation of H3-K9 and loss of H3-K4 methylation, which are early markers of X inactivation (11), were detectable in all cells with Eed localization to Xi, even at the morula stage (Fig. 2, E and F). Also, H4 hypoacetylation, a later marker for the X inactivation process (8, 11, 13), was seen underlying Eed foci at morula and subsequent stages (Fig. 2G). These results demonstrate that X inactivation initiates earlier than previously thought, at or even before morula stage, and that markers of X inactivation other than Eed-Ezh2 localization are established in ICM cells of early blastocysts.
Taken together, our results show that paternally imprinted X inactivation is established in all cells in early XX preimplantation embryos, implying that there is a subsequent reactivation event in cells of the ICM. Because Xp Xist expression is progressively extinguished in ICM cells during blastocyst maturation (6, 7), we infer that this may be the mechanism of X reactivation. This contrasts with differentiated somatic cells in which maintenance of X inactivation is Xist-independent (14, 15). Analysis of Eed-Ezh2 and H3-K27 methylation supports this interpretation. First, using immunoRNA-FISH (fluorescence in situ hybridization) analysis, we found that Eed-Ezh2 complexes dissociate rapidly from Xi as presumptive ICM cells begin to extinguish Xp Xist RNA expression (Fig. 3A). However, loss of tri-meH3-K27 staining on Xi disappeared more gradually. Thus, immunostaining for both Eed and meH3-K27 revealed cells in which Eed foci were lost, but meH3-K27 staining was still clearly detectable (Fig. 3B). In all instances, cells showing this pattern also had high overall levels of Eed staining in the nucleus, indicating that they represent ICM cells (Fig. 1B). In the ICM of mature blastocysts, both Eed and meH3-K27 staining of Xi were no longer detectable (Fig. 3C). At 5.5 days post coitum (dpc), as epiblast cells begin to undergo random X inactivation, localization of Eed-Ezh2 and associated tri-meH3-K27 were again detectable (Fig. 3D). This sequential loss and reestablishment—first of Eed-Ezh2 localization and then of tri-meH3-K27—provides a direct illustration of X reactivation in ICM cells.
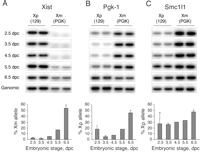
In light of evidence that chromosome-wide features of X inactivation are established in all cells of early embryos, we were interested to investigate allelic expression of individual X-linked genes. Embryos were obtained from crosses between Mus musculus domesticus and PGK strains, providing expressed single-nucleotide polymorphisms for quantitative allele specific assays (16). For early blastocyst and subsequent stages, XX and XY embryo pools were separated using a green fluorescent protein (GFP) transgene inherited on the paternal X chromosome (fig. S3A). We first analyzed Xist, which is expressed only from Xi. Consistent with previous studies, expression was exclusively from Xp during preimplantation development, with up-regulation of the Xm allele first detectable at the onset of random X inactivation at 5.5 to 6.5 dpc (Fig. 4A).
We then analyzed expression of the Pgk-1 gene (Fig. 4B), which is known to be subject to X inactivation, and also the Smc1l1 gene (Fig. 4C), which is located at the distal end of the X chromosome and is also subject to X inactivation (Fig. S3B). At 2.5 dpc (eight-cell to early morula stage), both genes were biallelically expressed, consistent with activity of Xp. However, Xp transcript levels were relatively low, particularly at the Pgk-1 locus. (In part, this may relate to the fact that both male and female embryos were included in the 2.5 dpc pool.) At 3.5 and 4.5 dpc (blastocyst stage), Xp Pgk-1 levels were further reduced to approximately 5%, whereas levels of Smc1l1 were relatively unchanged. In postimplantation embryos, Xp transcript levels increased again, and in embryonic ectoderm of 6.5 dpc XX embryos (after the onset of random X inactivation), the ratios of Xp and Xm alleles are essentially equivalent. Analysis of the Pgk-1 locus indicates inactivation of Xp begins as early as the eight-cell stage. The further marked reduction in Xp Pgk-1 RNA at the early blastocyst stage is consistent with inactivation occurring in all cells, as indicated by our analysis with chromosome-wide markers. In contrast, the Smc1l1 gene shows only partial X inactivation at the blastocyst stage, suggesting that the rate of X inactivation for individual genes may vary across the chromosome. Similar results, analyzing Pgk-1 and other X-linked loci, have been reported in previous studies (2, 17), leading to the idea that there may be a gradient of X inactivation in early embryos, centered on the Xist locus. In summary, our results demonstrate that imprinted X inactivation of the paternal X chromosome, assayed at the level of chromosome-wide histone modifications and also repression of at least some loci, occurs in all cells of early mouse embryos, and that it is then reversed selectively in ICM cells after extinction of Xp Xist RNA expression. This latter observation indicates that heritability of X inactivation in early embryos requires ongoing Xist RNA expression, unlike XX somatic cells in which loss of Xist has little or no effect (14, 15). Reversible Xist-dependent silencing has also been reported to occur in response to inducible Xist transgene expression in undifferentiated ES cells (18). Thus, our findings provide an in vivo corollary for this observation. Reversibility of facultative heterochromatin in early embryos and ES cells is mirrored in the capacity of these cell types to reactivate the X chromosome in a somatic cell nucleus in ES cell fusion hybrids (19) or after nuclear transfer (20). Indeed, our results help to understand these findings. First, repression of Xp Xist occurs specifically in Nanog-positive cells at the time they are first allocated, suggesting that this is a property inherent to the pluripotent ICM lineage. The same activity in ES cells could result in repression of the somatic Xi Xist allele in ES-somatic cell hybrids. This then would lead to X reactivation in the ES nuclear environment, where heritability of X inactivation is strictly Xist-dependent. In the case of nuclear transfer, the Xi from the donor somatic cell is also the Xi in TE and PE lineages, but random X inactivation occurs in the embryo proper (20). This would be explained again if repression of Xist occurs specifically in ICM cells. TE and PE lineages would inactivate in response to maintained expression of the somatic Xi Xist allele, whereas ICM cells would repress Xist, establishing the ground state for random X inactivation in the embryo proper.
Supporting Online Material www.sciencemag.org/cgi/content/full/303/5658/666/DC1 Materials and Methods Figs. S1 to S3 Table S1 References 17 October 2003; accepted 29 December
2003
This article has been cited by other articles:
Volume 303, Number 5658, Issue of 30 Jan 2004, pp. 666-669. Copyright © 2004 by The American Association for the Advancement of Science. All rights reserved. |

| ||

