 |
 |
Originally published in Science Express on 11 December 2003 Science, Vol 303, Issue 5658, 644-649, 30 January 2004
Epigenetic Dynamics of Imprinted X Inactivation During Early Mouse DevelopmentThe initiation of X-chromosome inactivation is thought to be tightly correlated with early differentiation events during mouse development. Here, we show that although initially active, the paternal X chromosome undergoes imprinted inactivation from the cleavage stages, well before cellular differentiation. A reversal of the inactive state, with a loss of epigenetic marks such as histone modifications and polycomb proteins, subsequently occurs in cells of the inner cell mass (ICM), which give rise to the embryo-proper in which random X inactivation is known to occur. This reveals the remarkable plasticity of the X-inactivation process during preimplantation development and underlines the importance of the ICM in global reprogramming of epigenetic marks in the early embryo. 1 CNRS UMR218, Curie Institute, 26 rue d'Ulm, Paris
75005, France. * To whom correspondence should be addressed. E-mail: edith.heard@curie.fr
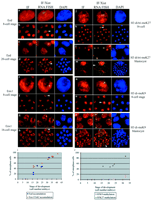
In mammals, dosage compensation between XX females and XY males is achieved by inactivating one of the two X chromosomes during early female embryogenesis (1). Classical biochemical and cytological analyses have suggested that in XX mouse embryos, there are three waves of X inactivation during development, correlating with the three earliest differentiation steps (2). In the first two lineages to differentiate, the trophectoderm and primitive endoderm of the blastocyst, which contribute to the extraembryonic tissues, X inactivation is subject to imprinting, with exclusive inactivation of the paternal X chromosome (Xp) (3, 4). This seems to be due to an imprint on the maternal X chromosome (Xm) to remain active, as well as a paternal imprint to inactivate (5). By the third wave of differentiation, in epiblast cells derived from the ICM, X inactivation is random, affecting either the Xp or the Xm (6, 7). Initiation of both imprinted and random X inactivation are dependent on a unique, untranslated RNA (Xist) that coats the X chromosome in cis and triggers its silencing (8, 9). In embryonic stem (ES) cells, which can recapitulate the random form of X inactivation upon in vitro differentiation, Xist RNA coating of the X chromosome is rapidly followed (within one to two cell cycles) by gene silencing (10). During early development, Xist is expressed from the two- to four-cell stage onward (11–13). Early Xist expression is exclusively of paternal origin (11); the maternal Xist allele is repressed until the morula stage (12). Despite the early Xist RNA coating of the Xp at the four-cell stage (12), the first cytologically detectable signs of X inactivation (a heteropycnotic, asynchronously replicating Xp) occur several cell divisions later, at about the 50-cell stage in the trophectoderm (14, 15). The reasons for this apparent delay, which contrasts with the rapidity of X inactivation that occurs after Xist RNA coating in differentiating ES cells, remain unclear. Furthermore, even though both the Xm and Xp are clearly active immediately after fertilization based on allozyme analysis (16–18), reverse transcription polymerase chain reaction (RT-PCR) studies have suggested that paternal alleles of some X-linked genes show lower transcriptional activity than their maternal counterparts during early embryogenesis (19, 20). Indeed, it has been suggested that a predisposition of the XP to inactivate may be carried over from its passage through the male germ line, where the Xp and the Y chromosome together form the highly condensed, heterochromatic sex vesicle, and this could underly imprinted X inactivation (21). Xist RNA and early chromatin changes on Xp. A number of unresolved questions thus surround the initiation and kinetics of imprinted X inactivation. We set out to address these questions with techniques that enable us to examine the status of the Xist RNA–coated Xp in individual cells during early preimplantation embryogenesis. Embryos from the two-cell to the blastocyst stage were isolated by flushing from the oviduct or uterus and analyzed directly, without in vitro culture, which can potentially lead to perturbations in epigenetic regulation. Using RNA fluorescence in situ hybridization (FISH), a domain of Xist RNA accumulation could be detected in all interphase blastomeres from the four-cell stage onward (Figs. 1 and 2 and fig. S1). These findings are consistent with previous studies (12, 13, 22, 23). We could detect a small punctate signal but no Xist RNA accumulation in a proportion of two-cell embryos (in the G2 phase based on developmental timing). At the four-cell stage, when paternal Xist RNA is just starting to accumulate, the Xist RNA domain was often small (Fig. 2 and fig. S3). We examined the early chromatin status of the Xp chromosome by means of Xist RNA FISH combined with immunofluorescence. In particular, we looked for Xp enrichment in H3 histones methylated at lysines 9 (K9) and 27 (K27) and for its association with the Eed and Enx1 polycomb group proteins (see table S1 for antibodies), because all of these marks have been shown to be characteristics of the X chromosome undergoing inactivation, and therefore represent candidates for the predisposition of the Xp to inactivate (24–27). Methylation of H3K9, in particular, has been reported to be associated with the Xp in the sex vesicle during spermatogenesis (28). When we examined four- and eight-cell stage embryos, no sign of either H3K9 and H3K27 methylation enrichment or of Eed and Enx1 accumulation could be detected on the Xist RNA–coated Xp (Fig. 1). At the 16-cell stage, however, these marks could be detected on the Xp in some blastomeres (Fig. 1 and fig. S2). Their similar time of onset on the Xp is consistent with Enx1 being the histone methyltransferase responsible for H3K27 methylation (26, 27). This is also the time window previously described for accumulation of the histone H2A variant, macroH2A, on the X chromosome (29), suggesting that major chromatin changes on the Xp begin at this stage. The proportion of blastomeres in which the Xp carried the Eed, Enx1, and H3K27 methylation marks varied considerably between embryos at the 16- to 32-cell stages (Fig. 1, i and r). By the blastocyst stage, however, the Xp carried these marks in the majority (>90%) of blastomeres. Methylation of H3K9 on the Xp showed different kinetics, with a later onset, which were first detected in embryos with >32 cells (Fig. 1 and fig. S2). By the midblastocyst stage, almost all trophectoderm cells had an Xp enriched in H3K9 methylation (Fig. 1, p and q). The later appearance of H3K9 methylation on the Xp suggests that this mark may be independently deposited, perhaps as a consequence of H3K27 methylation.
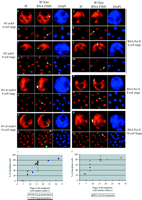
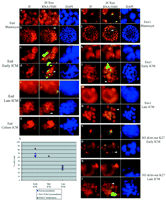
We also examined the timing of X-chromosome–wide hypoacetylation of H3K9 and hypomethylation of H3K4. These are early events during random X inactivation in differentiating ES cells; they occur within the same time window as H3K9 and K27 methylation (24, 26, 30). At the four-cell stage, no or very little depletion for these histone modifications could be detected at the site of Xist RNA accumulation (Fig. 2, a to d, and fig. S3A). However, at the 8-cell stage, both hypoacetylation of H3K9 and hypomethylation of H3K4 could be detected at the Xp in some blastomeres (Fig. 2, c to f), and by the 32-cell stage, the proportion of blastomeres with an Xp carrying these marks had risen to over 90% (Fig. 2i). Thus, hypoacetylation of H3K9 and hypomethylation of H3K4 occur after Xist RNA coating of the Xp chromosome, but precede Eed/Enx1 accumulation and H3K27 and K9 methylation. Transcriptional activity of the Xp. The surprisingly early appearance of these chromatin changes on the Xp suggested that the initiation of Xp inactivation could be more precocious than previously thought. Studies on the developmental timing of Xp inactivation have, in the past, involved analysis at the posttranscriptional level (RT-PCR) (19, 20) or posttranslational level [allozyme analyses, LacZ, and green fluorescent proteins (GFPs)] (4, 31, 32) and were hampered by the use of pools of embryos or cells, as well as the unknown half-lives of the X-linked transcripts and proteins assayed. We therefore decided to assess Xp silencing more directly, at the level of transcription and in single cells. Using an antibody (H5) that recognizes the elongating form of RNA polymerase II (RNA PolII), which is normally present only in transcriptionally engaged chromatin (33), we assessed the presence or absence of RNA PolII from the Xist RNA domain as a marker of transcriptional activity or silencing of the Xp. In differentiating female ES cells, we found that RNA PolII exclusion from the X chromosome is one of the earliest events in the X-inactivation process, after Xist RNA coating (34). Using this approach in preimplantation embryos, we could see no exclusion of RNA PolII at or around the location of the Xist pinpoint signal detected in two-cell, G2 stage embryos (fig. S3). The first signs of exclusion of RNA PolII from the site of Xist RNA accumulation on the Xp could be detected at the four-cell stage (Fig. 2, j to m). The number of blastomeres showing this pattern increased from one or two at the 4-cell stage to almost 100% by the 32-cell stage (Fig. 2r and fig. S3, i to l). The early initiation of transcriptional silencing of the Xp was confirmed with RNA FISH to detect nascent transcripts of the X-linked Chic1/Brx gene. At the two-cell stage, two Chic1/Brx signals, one of which was adjacent to the Xist pinpoint, could be seen in some embryos (fig. S3). At the 8-cell stage a Chic1/Brx RNA signal could be detected on the Xp within the Xist RNA domain in some blastomeres, but by the 16-cell stage, this pattern was never seen in more than one blastomere (35). Taken together, our data point to the initiation of transcriptional inactivation of the Xp from the four- to eight-cell stage, which is much earlier than previously thought. Before this, at the two-cell stage, the Xp seems to be active. Inactivation appears to be triggered by Xist RNA coating of the Xp and affects most if not all blastomeres by the 32-cell stage. Imprinted X inactivation thus initiates before any overt signs of cellular differentiation. Inactivation status of the Xp in the ICM. By the late morula/early blastocyst stage (the first sign of the blastocoel cavity), our findings suggest that essentially all blastomeres contain an inactive Xp, as assessed by RNA PolII exclusion, histone modifications, and Eed/Enx1 association. An important implication of this is that the Xp must be inactive even in the ICM, which gives rise to the epiblast. However, it is well known that random and not imprinted paternal inactivation occurs in this lineage. To resolve this paradox, we investigated the activity status of the Xp specifically in the ICM, using immunosurgery to isolate ICMs and eliminate all trophectoderm cells. When this procedure was performed on early blastocysts, the isolated ICM cells were found to contain a Xist RNA domain from which RNA PolII staining was excluded (fig. S4, e and f) and which was associated with H3K9 hypoacetylation, H3K4 hypomethylation, H3K9 and K27 methylation, and Eed and Enx1 accumulation (Fig. 3, k to n) (35). Thus, exactly as predicted by our kinetic analysis, the Xist RNA–coated Xp appears to be inactive in all cells, including those of the ICM, up to the early blastocyst stage. However, when ICM immunosurgery was performed on later (expanded or hatching) blastocysts, we found that, in contrast to trophectoderm cells, in the majority of ICM cells, Xist RNA appeared to be dispersed or absent and no Eed- or Enx-enriched domain could be found. Thus, during ICM growth, Xist RNA coating of the Xp is lost, and this is tightly coupled to loss of Eed and Enx1 accumulation on the Xp (Fig. 3h). However, the majority of these cells still contained what appeared to be an X-chromosome domain enriched in H3K27 methylation, even in the absence of Xist RNA coating. In ICMs from the latest blastocysts recovered, some cells had also lost the H3K9 and K27 methylation domain, even though these modifications could clearly be detected elsewhere in the nucleus (Fig. 3, q and r). Histone H3K9 and K27 methylation are therefore also lost on the Xp over time in the ICM, but after the dissociation of Xist RNA, Eed, and Enx1 (Fig. 4a).
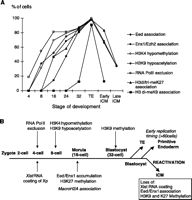
We also isolated the ICM from early blastocysts by immunosurgery and cultured them for 24 hours. In this procedure, the outgrowing cells are believed to be of the primitive endoderm type (15, 36). All outgrowing cells were found to have a Xist RNA domain that showed the chromatin characteristics observed in morulas and in the trophectoderm (Fig. 3g) (35), suggesting that the inactive state of the Xp is probably maintained upon differentiation of the primitive endoderm lineage. On the other hand, in cells at the center of the ICM, reversal of the inactive state (loss of Xist RNA coating, as well as loss of Eed and Enx association) could be detected, as in ICMs isolated directly from late blastocysts. Conclusions. We have made a number of findings concerning the process of X inactivation during early mouse development. We show that the imprint on the Xp is unlikely to be due to its global chromatin status but rather to the early activity of the paternal Xist gene. We also show that, although the Xp is initially active in early cleavage female embryos, it becomes rapidly inactivated after accumulation of Xist RNA at the four-cell stage, as assessed by the exclusion of RNA PolII, the absence of nascent X-linked transcripts, and the appearance of H3K4 hypomethylation and H3K9 hypoacetylation. Polycomb group proteins, which are believed to be involved in the early maintenance of the repressed state in several systems (37) and X inactivation in particular (26, 27), subsequently accumulate on the Xp and H3K27 methylation occurs. The variable time of onset we observed for these early maintenance marks over the 16- to 32-cell stage may explain the potential reversibility of Xp imprinted inactivation in some embryos, under abnormal circumstances such as in XpO (with no Xm and a single Xp) mice (38) or in androgenotes (XpXp) (22). From the 32-cell stage onward, however, we show that the Xp carries these epigenetic marks in virtually all cells of female embryos and maintains them in the trophectoderm and primitive endoderm. In these lineages, they are presumably further "locked in" by the shift in replication timing of the Xp (14). However, during ICM growth, we found that X inactivation is reversed. A rapid loss of Xist RNA coating of the Xp was observed, accompanied by the loss of Eed and Enx1 association, and this was followed by a more gradual loss of the H3 methylation marks, presumably by dilution of old nucleosomes through cell division. By the time implantation occurs, the Xp is no longer inactive and random inactivation of either the Xp or the Xm can occur in cells of the epiblast. The kinetics of these events are shown in Fig. 4a, together with the order of events during development (Fig. 4b). Our study reveals that the inactive state of the Xp and associated epigenetic marks are highly labile during preimplantation development. Their reversal in the ICM may be a reflection of more global reprogramming events occurring at this stage of development (39). The reasons for such a dynamic cycle of Xp inactivation, followed by reactivation, followed by random X inactivation in the mouse are unclear but could be due to a combination of evolutionary pressure to silence the Xp early in development, as predicted by the parental genome conflict theory (40), together with the efficient reprogramming activity of the ICM. Finally, in a study investigating X inactivation in cloned mouse embryos (41), a GFP transgene carried by a somatic cell–derived inactive X chromosome, was shown to be reactivated after transfer into an enucleated oocyte, but then became preferentially inactivated in extraembryonic tissues. In light of the results we present here, these findings are consistent with the idea that the presence of a transcribing Xist gene, whether paternally inherited or of somatic cell origin, is likely to be the only "imprint" required to trigger the chromatin modifications and epigenetic marks that lead to preferential X inactivation in the cleavage stage embryo.
Supporting Online Material www.sciencemag.org/cgi/content/full/1092727/DC1 Materials and Methods Figs. S1 to S4 Table S1 References and Notes 20 October 2003; accepted 1 December
2003
This article has been cited by other articles:
Related articles in Science:
Volume 303, Number 5658, Issue of 30 Jan 2004, pp. 644-649. Copyright © 2004 by The American Association for the Advancement of Science. All rights reserved. |

| ||

