RNAseq Anota
Trinity – micromamba do trinity
e Systems Biology
Primeira parte: MONTAGEM E ANÁLISE DE EXPRESSÃO
1° Passo: login usual como bioufmg@bioinfo.icb.ufmg.br
- acione o “environment”
conda assim: micromamba activate trinity
- entre em sua pasta com cd
sua_pasta
- mkdir
anotatrinity_aula
- cd anotatrinity_aula
cp -rf /home/treinamento/trinity_aula/* . [tem um ponto ai gente!]
- ls (o –rf ali em cima permitiu copiar as pastas rnaseq_data e trinity_saida)
- trinity já rodado gerou um “fasta” [less nele!], um “map” [less nele!] e uma pasta com os cálculoa trinity_saida [deixa quieta]. O comando abaixo sobrescreveria isso (rode opcionalmente):
---------------------------------------------------------------------------------------------------------------------------
Trinity --seqType fq --SS_lib_type RF --left rnaseq_data/Sp_log.left.fq.gz,rnaseq_data/Sp_plat.left.fq.gz
--right rnaseq_data/Sp_log.right.fq.gz,rnaseq_data/Sp_plat.right.fq.gz
--CPU 10 --max_memory 1G --output trinity_saida/
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
--left caminho de todas as reads no sentido forward, tanto log quanto plat
--right caminho de todas as reads no sentido reverse
--SS_lib_type (RF):
biblioteca paired-end
(Reverse+Forward)
--CPU (1): use 1 CPU
--max_memory (1G): Trinity irá utilizar 1GB de RAM
--output (trinity_saida/): diretório que Trinity vai criar para os cálculos
2° Passo: Análise do resultado, rode:
TrinityStats.pl trinity_saida.Trinity.fasta
- Tem menos genes que transcripts pois alguns tem isoformas (vide map)
Exemplo de N50 com transcrito 2 ocorrendo a 50% das bases ordenadas:
1111111111111111111111111222222222222222233333333333444444455555
3° Passo: Alinhe com Salmon os reads aos transcritos montados pelo Trinity (2 runs!!!!)
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg/micromamba/pkgs/trinity-2.15.1-pl5321hdcf5f25_4/bin/align_and_estimate_abundance.pl
--seqType fq --left
rnaseq_data/Sp_log.left.fq.gz --right rnaseq_data/Sp_log.right.fq.gz --transcripts trinity_saida.Trinity.fasta --est_method salmon --trinity_mode --prep_reference --output_dir salmon_saida/sp_log/
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg/micromamba/pkgs/trinity-2.15.1-pl5321hdcf5f25_4/bin/align_and_estimate_abundance.pl
--seqType fq --left rnaseq_data/Sp_plat.left.fq.gz
--right rnaseq_data/Sp_plat.right.fq.gz --transcripts trinity_saida.Trinity.fasta
--est_method salmon --trinity_mode --prep_reference --output_dir salmon_saida/sp_plat/
---------------------------------------------------------------------------------------------------------------------------
3° Passo: olhar o que o Salmon contou para cada gene
- cd salmon_saida
- cd sp_log
- less quant.sf.genes [uai, montagem DN42 é um artefato! Salmon contou zero reads!]
- cd ../../ para sair de sp_log e de salmon_saida de uma vez só
- DN48 bem expresso (alto TPM) e DN105 nem tanto, olhe DN75 em
4° Passo: comparando as fases log X platô – vc está na pasta original? Dê cd ../...
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg/micromamba/pkgs/trinity-2.15.1-pl5321hdcf5f25_4/bin/abundance_estimates_to_matrix.pl --est_method
salmon --out_prefix salmon_saida/salmon
salmon_saida/sp_log/quant.sf salmon_saida/sp_plat/quant.sf --gene_trans_map trinity_saida.Trinity.fasta.gene_trans_map
--name_sample_by_basedir
---------------------------------------------------------------------------------------------------------------------------
- cd salmon_saida [novamente pois tem arquivos novos]
- less salmon.gene.counts.matrix [tem gene mais expresso em log, igualmente expresso...]
- less salmon.gene.TMM.EXPR.matrix [TMM tira o efeito dos genes superativos no tecido]
5° Passo: usando edgeR ver o que é ou não significativamente diferente, volte à sua pasta
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg/micromamba/pkgs/trinity-2.15.1-pl5321hdcf5f25_4/opt/trinity-2.15.1/Analysis/DifferentialExpression/run_DE_analysis.pl --matrix salmon_saida/salmon.isoform.counts.matrix --method
edgeR --output edgeR_saida/ --samples_file samples.txt --dispersion 0.1
---------------------------------------------------------------------------------------------------------------------------
- --samples_file (samples.txt): um arquivo que descreve as replicatas versus condições dos seus experimentos:
-
#condition #samples
-
condA repA1
-
condA repA2
-
condB repB1
-
condB repB2
- --dispersion (0.1): parâmetro utilizado na função binomial negativa do edgeR para avaliar as contagens dos genes e ver se a diferença é significativa.
6° Passo: veja o que foi criado em edgeR_saida: cd edgeR_saida
.DE.results: resultado incluindo o fold change e a significância estatística (FDR), dê less:
less
salmon.isoform.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results
.MA_n_Volcano.pdf: nossos gráficos juntos, sendo um de MA e um Volcano!
Para ficar facilitar a visualização no navegador, digite um comando para simplificar o nome dele e copiar ele na sua pasta (um diretório acima) assim:
---------------------------------------------------------------------------------------------------------------------------
cp salmon.isoform.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results.MA_n_Volcano.pdf
../MA_Volcano.pdf
---------------------------------------------------------------------------------------------------------------------------
Abra agora uma nova página no seu navegador no link:
http://bioinfo.icb.ufmg.br/bioufmg/look/ login (bioufmg) e senha (carrarataxis).
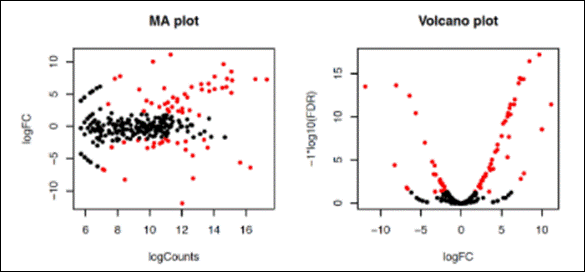
Ao abrir o
arquivo PDF (MA_Volcano.pdf) podemos
verificar que as contagens com um valor de FDR < 0.5 (os bãos de olhar) estão coloridas de vermelho. Fold change expressa o número de
dobradas ou caídas pela metade: dobrar é 1, 2 é quadruplicar...
7° Passo: usando awk para selecionar valores de FDR (na pasta edgeR-saida)
---------------------------------------------------------------------------------------------------------------------------
cat
salmon.isoform.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results | awk '$7 <= 0.05 {print $1,$4,$7}' | less
---------------------------------------------------------------------------------------------------------------------------
- cat vc sabe joga na tela o arquivo com nome grande
- awk é o comando legal para analisar tabelas
- a ‘aspas simples’ abre a condição, no caso coluna 7 (FDR) menor que 5%
- usamos $1$4,$7 para ver o Fold Change (dobramentos) da coluna 4 e FDR da coluna 7
- troque less por wc –l para contar quantas linhas têm diferença com FDR menor que 5%:
---------------------------------------------------------------------------------------------------------------------------
cat
salmon.isoform.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results | awk '$7 <= 0.05 {print $1,$4,$7}'
| wc -l
---------------------------------------------------------------------------------------------------------------------------
8° Passo: votle à sua pasta com cd .. e rode sh filtrar.sh
Vamos pegar todas as sequências de todos os genes que possuem o FDR <= 0.05 utilizando o script filtrar.sh com sh filtrar.sh
DE_genes.txt: contém o nome de cada gene diferenialmente expresso;
DE_genes.fasta: contém o cabeçalho e a sequência do gene (o fasta);
2° PARTE: ANOTAÇÃO DOS GENES
DIFERENCIALMENTE EXPRESSOS
9° Passo: conte quantos genes estão diferencialmente explressos
---------------------------------------------------------------------------------------------------------------------------
cat DE_genes.fasta | less
cat DE_genes.fasta | wc
---------------------------------------------------------------------------------------------------------------------------
10° Passo:
Na sua pasta vamos anotar os fastas de DE_genes.fasta com Diamond
---------------------------------------------------------------------------------------------------------------------------
./Diamond blastx -d uniprot_sprot.dmnd
-q DE_genes.fasta -v --outfmt
6 -e 1e-10 --max-target-seqs 1 --outfmt
6 -o blastx.saida -b4
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
- Estamos rodando o blastx do Diamond, uma versão acelerada programaticamente;
- -d (uniprot_sprot.dmnd): base de dados SwissProt-Uniprot;
- -q (DE_genes.fasta): indicamos a query, os transcritos diferencialmente expressos
- -v: modo verbose, para poder mostrar letrinhas no terminal;
- -e (1e-10): valor de e-value de corte;
- --max-target-seqs (1): para poder anotar somente o best-hit de cada transcrito;
- --outfmt (6): define o modelo do qual será o arquivo de saída, que neste caso, é igual ao formato tabular de saída do programa BLAST;
- -o (blastx.outfmt6): arquivo de output.
- -b4: argumento para poder não limitar a memória no uso do Diamond.
11° Passo: ls e less blastx.saida
- Coluna 1: Nome da query;
-
Coluna 2:
Nome da referência encontrada pelo
transcrito (hit);
- Coluna 3: porcentagem de identidade;
- Coluna 4: tamanho do alinhamento;
- Coluna 5: número de mismatches;
- Coluna 6: número de gaps;
- Coluna 7; posição de início na sequência da query;
- Coluna 8: posição final na sequência da query;
- Coluna 9: posição de início na sequência da referência;
- Coluna 10: posição final na sequência da referência;
- Coluna 11: e-value;
- Coluna 12: bit score.
12° Passo: separando a coluna dos hits com cut -f 2 [field 2 = coluna 2]
cat blastx.saida | cut -f 2
Selecione todas as linhas contendo estes nomes, o PuTTy já copia ao iluminar
13° Passo:
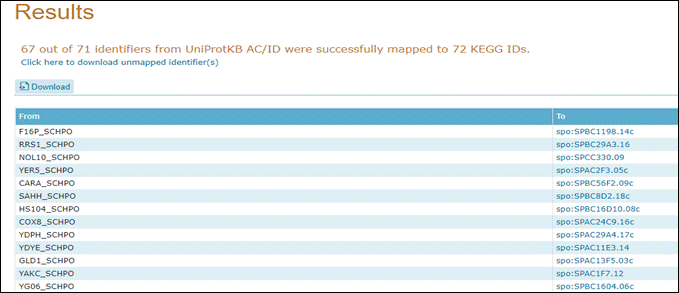
Acesse agora, numa nova aba do seu navegador, o site uniprot.org.
Logo no cabeçalho do site vamos clicar em ID mapping:
Cole o texto que você copiou do PuTTY (ctrl + v) em Enter one of more IDs;
Selecione, na parte de To database::
To Genome Annotation Databases >> KEGG. Por fim, clique em Map.
Clique em Completed e acesse a tabela como abaixo
Expore as proteínas que têm Pathway: a sexta: SPBP35G2.12; a oitava: SPAC6G9.09c; a nona: SPAC13F5.03c e a décima: SPCC1739.13. Essas todas tem Pathway pra vc olhar e as proteínas vão estar em vermelho nos Pathways! Aqui uma via da nona SPAC13F5.03c uma glycerol dehydrogenase de EC number 1.1.1.6
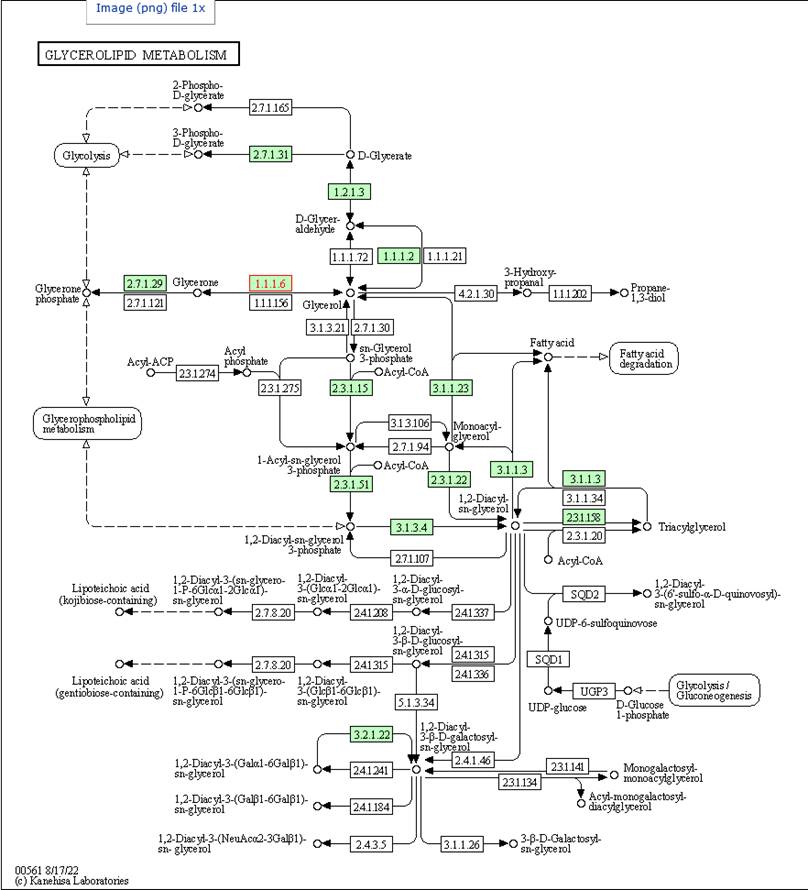
Agora você pode explorar todas as vias e genes que você identificou como diferencialmente expressos.
14° Passo:
Outra maneira de explorar genes diferencialmente expressos é a biologia de sistemas, Entre no site do String: . https://string-db.org/ e clique em SEARCH
Opte na barra lateral por Multiple proteins
Indique o organismo Schizosaccharomyces pombe e SEARCH
Aceite a conferência de entradas (CONTINUE) e veja a rede de interações PPI
Prefira trocar
em Settings
para High confidence 0.7 mas hj use 0.4
Repare que ele mostra vários tipos de interações, Textmining, Experiments...
Peça para mostrar não mais que 10 interações com a first shell da base de dados e UPDATE e depois não mais que 10 interações com a second shell e UPDATE
Veja em Analysis as vias mais enriquecidas
Teste o Clusters com MCL e inflation 3 e APPLY, selecione o maior cluster e veja o que ele faz (clique nele)
Volte em Settings e deixe só as arestas de dados experimentais
Foram 2 exemplos
de análise de biologia de sistemas: Pathways e Redes.
Como a aula foi preparada:
- download de micromamba:
curl
-Ls
https://micro.mamba.pm/api/micromamba/linux-64/latest | tar
-xvj bin/micromamba
- instalação de micromamba:
./bin/micromamba shell init -s bash -p ~/micromambasource ~/.bashrc
micromamba create -n trinity trinity R -c conda-forge –c bioconda
- ativação do env
trinity micromamba:
micromamba activate trinity