Aula
– Montagem de genomas
1. Abra um terminal em sua máquina Linux e crie uma pasta mkdir aula_montagem e entre nela cd aula_montagem
2. Agora abra uma conexão com a bioinfo ssh bioufmg@bioinfo.icb.ufmg.br
3. Entre em sua pasta cd eusoujacu crie um diretório
mkdir aula_montagem e entre nela cd aula_montagem
4. Copie o material para essa pasta cp /home/treinamento/velvet_aula/* . (olha o ponto!)
5. Dá um ls e veja que você tem três arquivos,
dois arquivos de sequenciamento (reads_forward.fastq
e reads_reverse.fastq) e um arquivo com o genoma de
referência (genoma.fasta)
6. Primeiro vamos olhar a qualidade dos reads com o programa fastQC, ele vai gerar várias saídas. Para ver as saídas mais rapidamente veremos o resultado em bioinfo.icb.ufmg.br/bioufmg (user bioufmg e senha invertida). Na bioinfo http://bioinfo.icb.ufmg.br/bioufmg abra o link olhe, que mostra na web o conteúdo de
todas as pastas (user bioufmg e senha invertida) em bioufmg. Assim, toda figura gerada
lá pode ser vista com o navegador!
7. Para observar a qualidade das bases nos arquivos reads_forward.fastq
e reads_reverse.fastq, crie o diretório sem_trim e execute o script fastqc
(com –t especifique rodar com um único core/thread)
mkdir sem_trim (o
programa exige que o diretório de output seja criado antes...)
fastqc -o sem_trim -t 1 reads_forward.fastq reads_reverse.fastq
8. Veja a saída abrindo
sua pasta (no navegador) e seguindo o caminho: aula_montagem/fastqc_output/reads_forward_fastqc/fastqc_report.html
para informações dos reads forward
e
aula_montagem/fastqc_output/reads_reverse_fastqc/fastqc_report.html para
informações dos reads reverse
9. Vamos filtrar esses reads com Trimmomatic assim:
java -jar /opt/treinamento/trimmomatic.jar PE -phred33 reads_forward.fastq reads_reverse.fastq trimmed_forward_paired.fastq trimmed_forward_unpaired.fastq trimmed_reverse_paired.fastq trimmed_reverse_unpaired.fastq LEADING:15 TRAILING:15 SLIDINGWINDOW:4:15 MINLEN:36
Os parâmetros para trimming são:
LEADING: Remove bases com baixa qualidade no início da sequencia (qualidade
abaixo de 3)
TRAILING: Remove bases com baixa qualidade no final da sequencia
(qualidade abaixo de 3)
SLIDINGWINDOW: Utiliza uma janela deslizando com 4 bases e corta quando
a qualidade média é menor que 15
MINLEN: Remove sequencias menores que 36 bases
10. Agora refaça o processo do fastQC com os reads filtrados criando diretório com_trim e
fastqc -o com_trim -t 1 trimmed_forward_paired.fastq
trimmed_reverse_paired.fastq
Veja o report do programa na sua pasta no navegador, em bioinfo.icb.ufmg.br/bioufmg/you@bioinfo procure:
trimmed_forward_paired_fastqc/fastqc_report.html
trimmed_reverse_paired_fastqc/fastqc_report.html
Melhorou?
11. Com os reads trimados
fazer a montagem sem referencia (do zero) com o programa Velvet
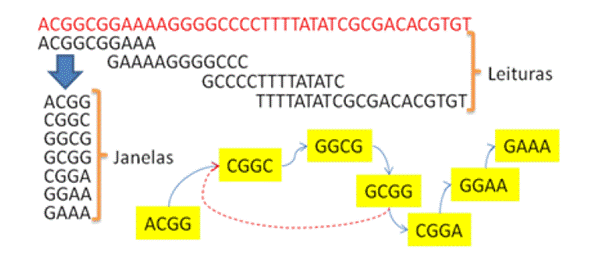
12. Rode o velvet assim:
Crie os índices (o parâmetro numérico refere-se ao tamanho do k-mer utilizado, sempre números ímpares)
velveth montagem 31 -fastq
-shortPaired -separate trimmed_forward_paired.fastq
trimmed_reverse_paired.fastq
Execute a montagem
velvetg montagem/ -exp_cov auto -scaffolding
yes
Após a execução, o Velvet apresenta algumas informações da montagem. Os nodes
são o número de contigs gerados. O n50 diz que metade do genoma está
representada em contigs maiores que esse valor.
![]()
13. Verifique o
arquivo de montagem
cd montagem
less contigs.fa
14. Agora vamos fazer o outro tipo de montagem que é "com genoma de
referencia" usando o programa Bowtie.
Primeiro volte ao diretório aula_montagem
cd ..
Rode o bowtie assim:
Crie um bowtie index usando o genoma de
ancoragem (genoma.fasta) fornecido:
bowtie2-build genoma.fasta
genoma.build
Alinhe os reads no index do genoma de
referência (genoma.build), criando um arquivo de alinhamentos reads_mapeados.sam no formato chamado SAM (-S manda criar o
SAM)
bowtie2 -x genoma.build
-1 reads_forward.fastq -2 reads_reverse.fastq
-S reads_mapeados.sam
15. Para ver o resultado teremos que abrir um programa Java na sua
máquina Linux (ou na windows)
chamado Tablet. Você pode rodar ele da web usando este link. Ele vai abrir
um ambiente gráfico. Se não funcionar na sua máquina pessoal, você pode fazer o
download do programa aqui.
16. Temos que pegar o arquivo de saída do Bowtie
(reads_mapeados.sam) e o genoma de referência
(genoma.fasta) e trazer para a sua máquina. O melhor a fazer
é achar o resultado usando o link olhe (link
bioufmg e inverta a senha ou pergunte) e "copiar link", depois salvar link como, com o botao da direita.
17. Memorize onde salvou os arquivos e vamos abrir pelo
modo gráfico do Tablet. 18. Aberto o arquivo ".sam" de saída do Bowtie e o genoma.fasta de referência, você vai ver um monte de reads ancorados na
referência. Clique em: "Open Assembly"
Na primeira janela selecione o arquivo: “reads_mapeados.sam” Na segunda janela selecione o genoma de referência: “genoma.fasta” Clique em open. 19. Não é aula de transcriptômica, então vamos aprender a exportar os
resultados do Bowtie usando o SAMTools.
De volta na pinguim: Converta o arquivo SAM para a versão binária BAM samtools view -S reads_mapeados.sam -b -o reads_mapeados.bam Ordene os reads samtools sort reads_mapeados.bam -o reads_mapeados_ordenados.bam Crie um índice para o BAM samtools index
reads_mapeados_ordenados.bam Crie os contigs com o consenso do mapeamento com samtools, bcftools, que manipula VCF (Variant Call Format, texto) e BCF (VCF tornado binário) e um script em perl vcfutils.pl para montar um .fastq samtools mpileup -ABuf genoma.fasta reads_mapeados_ordenados.bam | bcftools
call -c | vcfutils.pl vcf2fq > montagem_por_referencia.fastq
- pode dar uma olhadinha no .fastq gerado com less 20. Converta o arquivo fastq para fasta seqtk seq -a
montagem_por_referencia.fastq > montagem_por_referencia.fasta Visualize a montagem less montagem_por_referencia.fasta